Main Menu (Mobile)- Block
Main Menu - Block

The Transcription Imaging Consortium (TIC) (2006-2017) developed tools for in vitro and in vivo single molecule imaging and live-cell microscopy which enabled the investigation of the assembly, dynamics, and functional specificity of the eukaryotic transcription machinery.
Organisms depend on finely tuned gene expression for their development and survival. Despite sharing a single genome, the cells of an organism develop their own identity by activating unique sets of genes, calling on a large collection of regulators to precisely modulate the expression level of each one. Gene activity is carefully regulated according to cell type, developmental stage, and ever-changing environmental conditions.
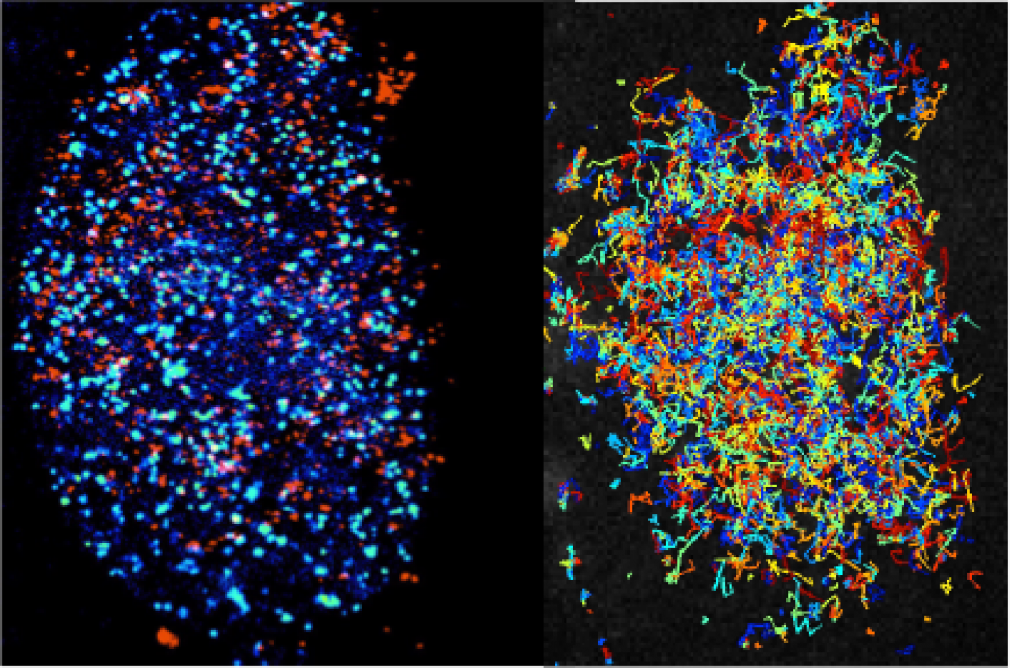
Transcription, which generates a RNA copy of the information encoded in a gene so that it can be used to produce a specific protein, is the first step of gene expression. Transcription of a single RNA molecule requires the coordinated activity of a vast number of factors, many of which interact and dissociate in less than a second. These events vary between cells, and individual cells can display highly variable gene expression profiles.
Many fundamental questions about how gene expression is regulated in eukaryotic cells remain largely inaccessible with existing tools. To observe individual molecules of DNA interacting with the proteins that initiate, carry out, and regulate transcription, we need better imaging technologies, including both microscopes and labeling reagents.
The Transcription Imaging Consortium, which formed in 2006 as one of Janelia's first collaborative projects, brought together individuals with expertise in biophysics, cell biology, superresolution imaging, biochemistry, and chromatin biology to develop new tools for the study of the assembly, dynamics, and function of the eukaryotic transcriptional machinery. A central part of our mission was to actively share our output with the scientific community.
We focused on the following areas:
- Instrumentation Technology Development
- Integrating Technologies to Image Multiple Stages of the Transcription Cycle
- Imaging Chromatin Architecture and Long Range DNA Interaction Dynamics
- Quantifying Transcription Regulation across Genes Networks in Cells and Tissue
To find out more, please visit our Research page.