Main Menu (Mobile)- Block
- Overview
-
Support Teams
- Overview
- Anatomy and Histology
- Cryo-Electron Microscopy
- Electron Microscopy
- Flow Cytometry
- Gene Targeting and Transgenics
- Immortalized Cell Line Culture
- Integrative Imaging
- Invertebrate Shared Resource
- Janelia Experimental Technology
- Mass Spectrometry
- Media Prep
- Molecular Genomics
- Primary & iPS Cell Culture
- Project Pipeline Support
- Project Technical Resources
- Quantitative Genomics
- Scientific Computing Software
- Scientific Computing Systems
- Viral Tools
- Vivarium
- Open Science
- You + Janelia
- About Us
Main Menu - Block
- Overview
- Anatomy and Histology
- Cryo-Electron Microscopy
- Electron Microscopy
- Flow Cytometry
- Gene Targeting and Transgenics
- Immortalized Cell Line Culture
- Integrative Imaging
- Invertebrate Shared Resource
- Janelia Experimental Technology
- Mass Spectrometry
- Media Prep
- Molecular Genomics
- Primary & iPS Cell Culture
- Project Pipeline Support
- Project Technical Resources
- Quantitative Genomics
- Scientific Computing Software
- Scientific Computing Systems
- Viral Tools
- Vivarium
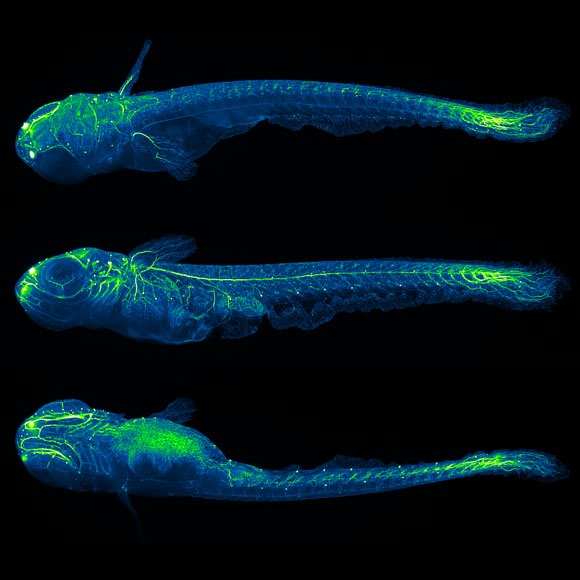
Our goal is to understand the principles underlying the formation of the nervous system. We use light-sheet microscopy and computer vision to quantitatively study development and emergence of function in the early nervous system of fruit fly, zebrafish and mouse.
Lab Updates
Animal development is one of the most complex processes encountered in biology. In early embryonic development of vertebrates and higher invertebrates, a single cell is transformed into a fully functioning organism comprising tens of thousands of cells that build tissues and organs able to perform the most challenging tasks. Understanding development and the concurrent emergence of function at this system-wide level is one of the most fundamental goals of biology.
Working toward this goal, we perform highly interdisciplinary research at the interface of neuroscience, developmental biology and biophysics, and develop high-speed light-sheet microscopy technology and automated approaches to computer vision to enable this research. The objective of our research is to uncover the fundamental rules governing neural development, and to systematically link development to the functional activation of circuits in the nervous system. In the long-term perspective, we would like to use these data to establish and validate a computer model of the developing nervous system and, ultimately, of the entire embryo.
To elucidate these key principles at the system level, we (1) perform live imaging of entire developing fruit fly, zebrafish and mouse embryos, focusing in particular on the developing nervous system, (2) computationally analyze the patterns of cell migration, cell division and axonal outgrowth underlying the formation of the nervous system, and (3) study the emergence of functional connectivity and patterned neural activity in the early nervous system.