Main Menu (Mobile)- Block
Main Menu - Block

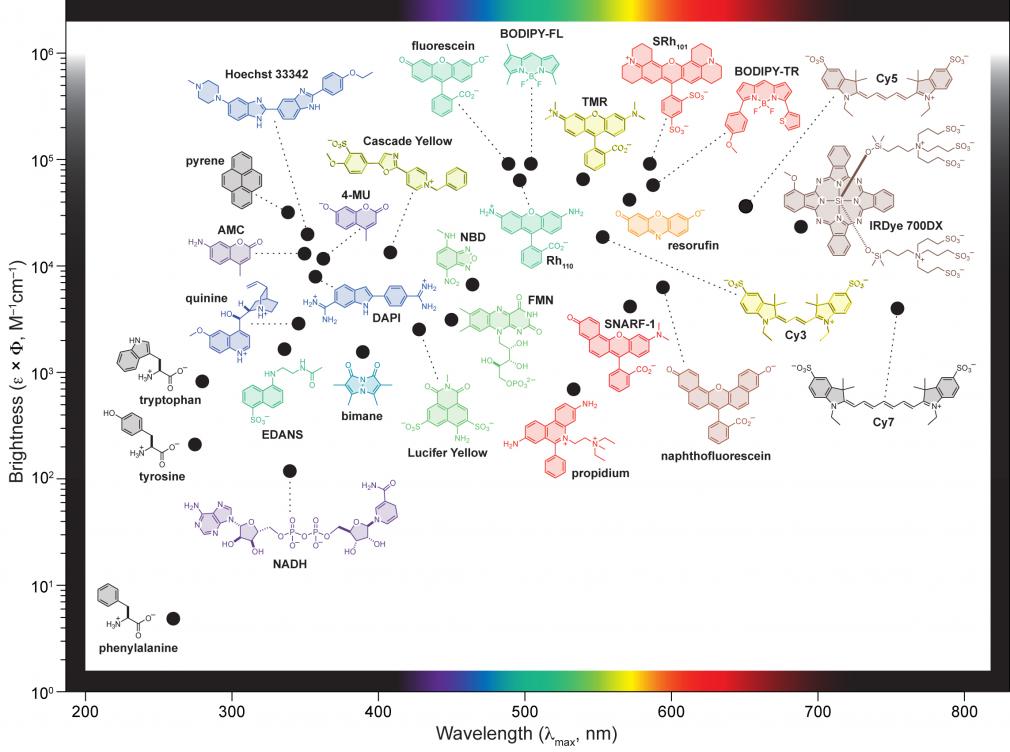
We work at the interface of chemistry and biology, assembling small molecule fluorescent dyes that facilitate sophisticated biological studies.
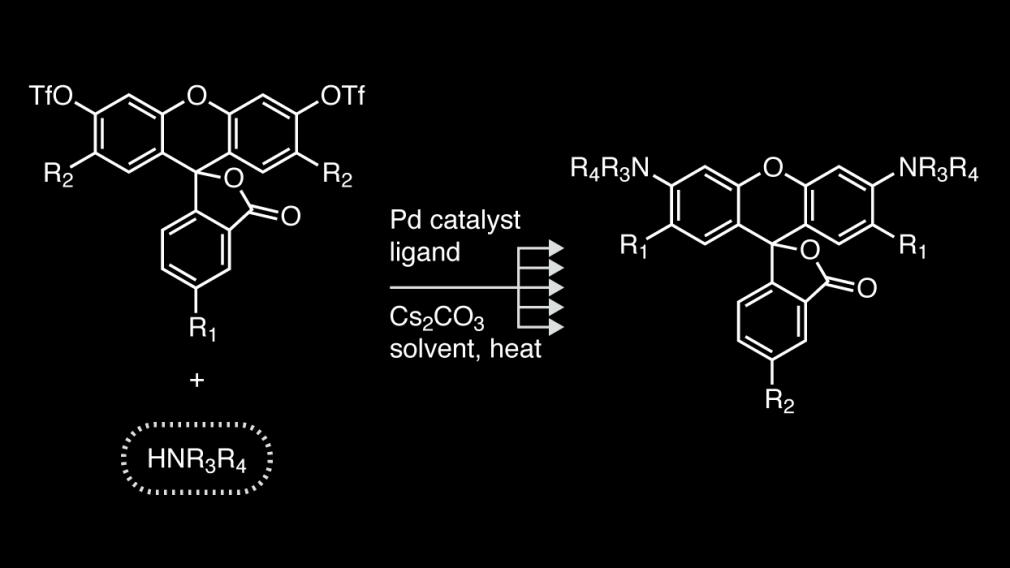
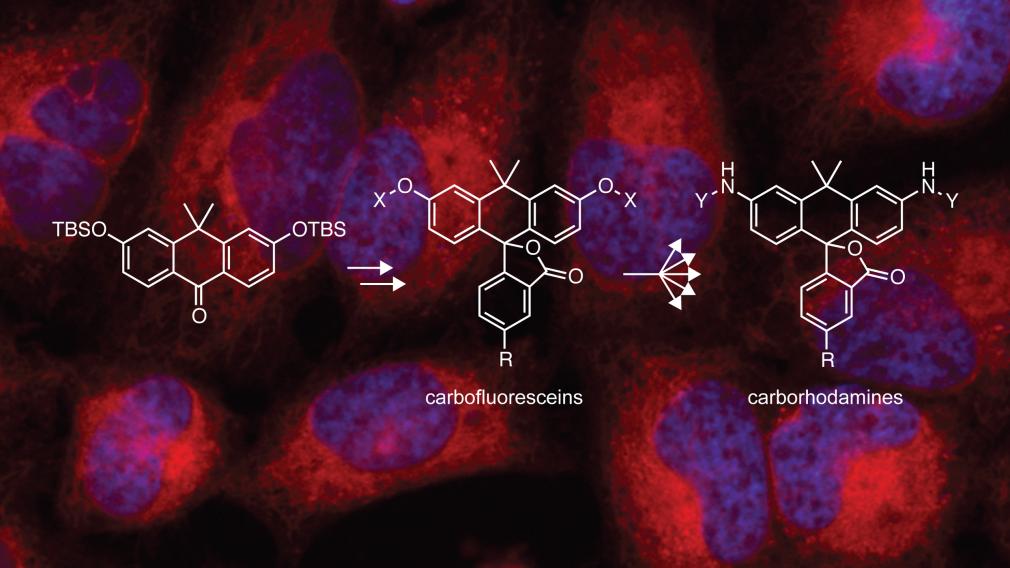
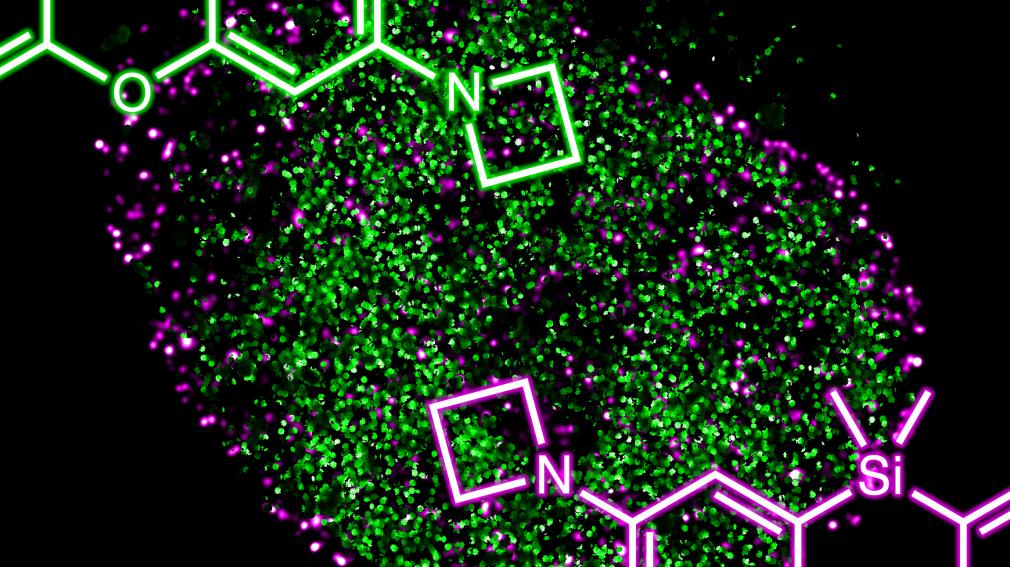
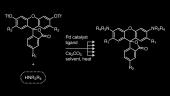
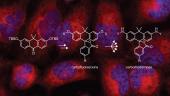
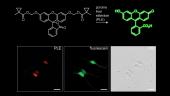
We are interested in designing and building small molecules to measure or manipulate biological systems. Our laboratory synthesizes bright fluorescent labels that enable the imaging of individual molecules in living cells. We also develop fluorogenic probes where the chemical and photophysical properties can be masked by assorted molecular functionalities and then unmasked by a user-designated process involving light, enzymatic activity, or environmental changes. This chemical masking suppresses unwanted fluorescence signals, thereby functioning as a filter for bioimaging and other experiments. Combining these novel compounds with advances in instrumentation, protein engineering, and genetic manipulation allows us to devise sophisticated ways to illuminate complex biological systems.
We use modern organic chemistry to transform old dyes into tools for 21st century biology.
-Luke Lavis