Main Menu (Mobile)- Block
Main Menu - Block

How have genes and neural circuits evolved to generate diversity of Drosophila courtship?
How do aphids hijack plant development to induce galls?
Lab Updates
This is an extraordinary time to study the origins of biological diversity. Many deep and old evolutionary questions are being solved by the application of new technologies.
We develop new approaches and new tools to ask how molecular changes cause phenotypic evolution, especially the evolution of behavior. Also, we recently started a new project in the lab, studying how aphids induce dramatic transformations of plant tissue into nutritious, protective homes called galls.
Evolution of Behavior
We aim to identify the genetic and neural basis for the evolution of behavior, because we believe that there may be general principles to behavior evolution, similar to the principles that have been revealed for morphological evolution (Stern & Orgogozo, 2009; Stern 2010). One major goal is to identify the individual molecular changes that have contributed to evolutionary change, since we believe that these “quanta” of evolution provide the deepest insight into the evolutionary process.
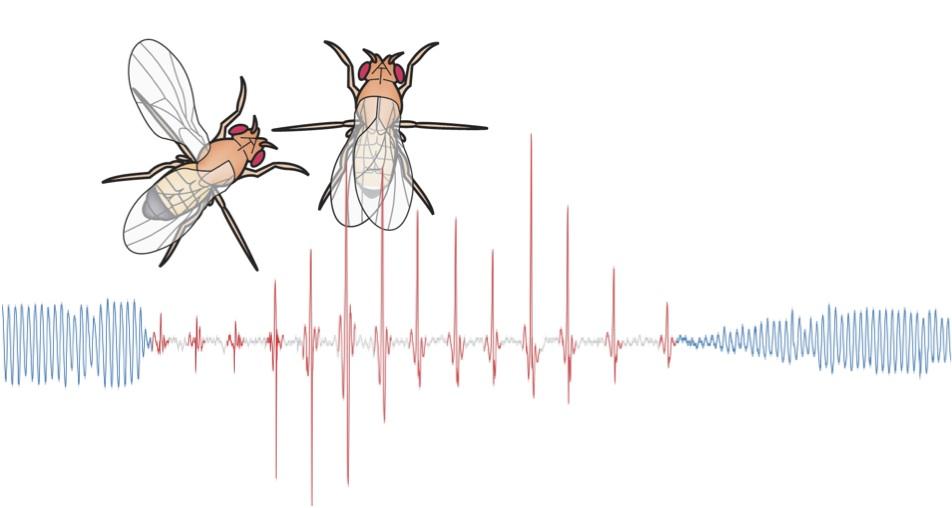
Our behavior studies focus on the evolution of Drosophila male courtship song, a rapidly-evolving behavior. We have developed multiple tools to aid these studies, including high-throughput genotyping (Andolfatto et al. 2011) and phenotyping platforms (Arthur et al. 2013; Arthur et al. 2021), and new transgenic reagents for multiple species (Stern et al. 2017; Stern 2022).
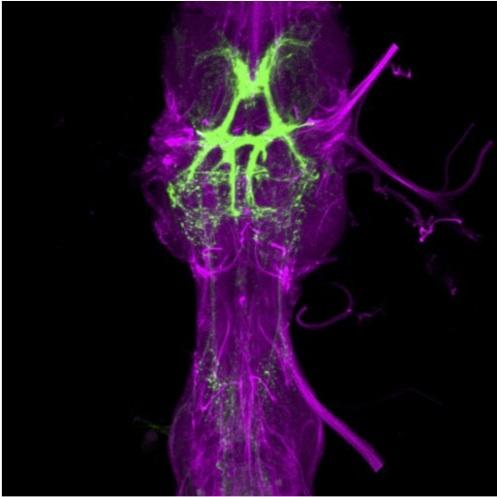
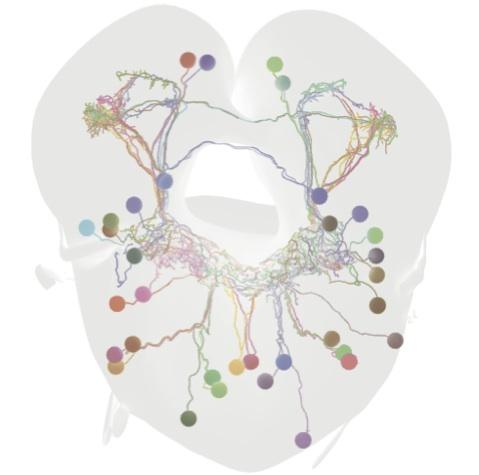
Using these tools, we have identified a mutation generating natural variation in courtship song (Ding et al. 2016) and begun to examine how individual homologous song neurons contribute to divergent songs in different fly species (Ding et al. 2019).
We continue to develop new tools and reagents to facilitate efficient discovery of the genetic and neural causes of behavior evolution.
Mechanisms of gall induction
Thousands of arthropods manipulate plants to induce dramatic “organs” called galls that the arthropods exploit for nutrition and protection. The mechanisms that arthropods use to exploit plant development remain largely unknown.
We have focused on aphids of the species Hormaphis cornu, which induce prominent “cone galls” on the leaves of witch hazel, Hamamelis virginiana. This is an entirely wild system and would be challenging to bring into the laboratory, since the aphids migrate annually between two trees, Hamamelis and River Birch (Betula nigra). Fortunately, both trees can be found on Janelia Research Campus. We have applied genomic approaches and genetic mapping studies to this large, local population and implicated a large, diverse family of novel proteins, encoded by what we call bicycle genes, as potential regulators of gall development (Korgaonkar et al. 2021). (We call these bicycle genes because the proteins encoded by these genes tend to carry a tandem pair of Cysteine-Tyrosine-Cysteine — or, abbreviated, CYC — motifs; thus, bi-CYC-like proteins. We have recently discovered that the proteins encoded by some genes of this family carry one, three, four, or many CYC domains and we call these unicycle, tricycle, tetracycle, and megacycle genes (Stern & Han.2022).)
Study of bicycle genes presents many opportunities and challenges. Amongst the opportunities, we have recently discovered that all aphids, and their sister group scale insects, carry bicyclegenes, suggesting that this a widespread class of effector proteins used by all aphids and scale insects to manipulate host plants (Stern & Han.2022). We are therefore now studying bicyclegenes in both gall-forming and non-gall-forming aphids.
Amongst the challenges are that predicted Bicycle proteins do not have obvious homology to other proteins of known function. Thus, their molecular mode of action cannot be predicted using bioinformatic approaches. We are therefore embarking on biochemical and genetic studies of Bicycle protein functions. Stay tuned!
"I came to Janelia to do science with my own hands. I am an inveterate lab rat and I really appreciate the fact that I can spend all day, every day, doing experiments that enhance the lab's research. I feel incredibly lucky to be a post-doc in my own lab!"
David L. Stern, December 2014
"Throughout their careers [Thomas Hunt] Morgan and [his] students worked at the bench. The investigator must be on top of the research if he or she is to recognize unexpected findings when they occur."