Movie 1. iPALM 3D rendering of U2OS cell expressing αv integrin tagged with tdEos. In this rendering, the lumen of the endoplasmic reticulum (ER) is clearly visible. Taken from Shtengel, et al., Proc. Nat. Acad. Sci. 106(9): 3125-3130 (2009).
Movie 2. iPALM 3D rendering of U2OS cell expressing αv integrin tagged with tdEos. In this rendering, the lumen of the endoplasmic reticulum (ER) is clearly visible. Z-position is color coded such that red is nearest the coverslip, while blue is furthest away. Taken from C. Henry Arnaud, Chem. Eng. News, 87 (36), pp 69–71 (2009)
Applications
⦁ Because of iPALM’s excellent resolution capability, particularly in the axial direction, it lends itself well to studies of protein interaction, characterizing cellular ultra-structure, as well as protein-plasma membrane associations, among other studies.
⦁ Demonstrations of cytoskeletal morphology, focal adhesion complex arrangement, endoplasmic reticulum (ER) structure, mitochondrial physiology have been made.
Strengths
⦁ 2-5x higher lateral resolution compared to other super-resolution techniques such as SIM
⦁ 2-5x axial resolution improvement compared to other commercially available STORM/PALM systems based on astigmatic detection. In fact, the iPALM is currently the localization-based super-resolution microscope with the most accurate z-resolution.
⦁ Lower excitation intensities than STED (kW/cm2 vs. MW-GW/cm2), thus limiting photodamage
⦁ Multi-color (sequential) imaging
⦁ Sample geometry (sealed sample chamber) is compatible and beneficial for both photoactivatable (endogenous) protein labels and dSTORM (exogenous) labels.
Basic Principles
Interferometric photoactivation and localization microscopy (iPALM) is a novel technique that combines single molecule localization (SML) with multi-phase interferometry. Because it offers unparalleled spatial, nearly isotropic resolution in the lateral and axial directions, to well below the diffraction limit (<25nm), it is suitable for studies that require near electron microscopy levels of resolution, but within intact, fluorescently labeled samples. The iPALM technique is particularly advantageous due to its high axial resolution, which has been demonstrated to exceed that of other super-resolution methods based on biplane/multifocal detection, as well as “engineered” point spread function techniques including astigmatic approach. To achieve such high spatial resolution, however, the iPALM is more limited in the available temporal resolution than other super-resolution modalities such as SIM. Thus, fixed samples are most often used in this approach.
To achieve such resolution, this approach makes use of single molecule localization (SML). This principle relies on two key aspects: First, via the use of photoswitchable and/or photoactivatable fluorescent proteins or dyes in combination with special buffer formulations, samples can be imaged such that only a small, random subset of its fluorophores are visible at any given time. By tuning the excitation laser power, as well as a UV wavelength “activation” laser, images can be acquired that contain only single, isolated fluorophores. Second, based on the assumption that acquired images contain individual, well-isolated fluorophores, their lateral location can be found with much higher precision than what is possible using normal diffraction-limited microscopy by computing the centroid of the detected point-spread function. By repeatedly imaging a sample over time, the nanoscale location of nearly all fluorophores can be found. Figure 1 illustrates these concepts.
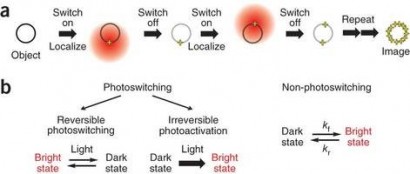
Figure 1. Basic principle of single molecule localization (SML) microscopy. Adapted from Dempsy et al., Nat. Meth. (2011)
To achieve its unique axial (z) resolution, iPALM makes unique use of interferometry principles that distinguish it from other commercially available SML approaches. In classical interferometry, a coherent beam of light can be split into two or more paths, and then recombined to form an interference signal. The features of this signal can be used to compute the difference path length to within nanometers or less. To adapt this principle to iPALM, we make use of the fact that a single fluorophore is in fact a quantum source – that is, an emitted photon from a single fluorescent molecule can interfere with itself. In order to provide two paths for the fluorescent signal to traverse, iPALM makes use of two opposing (top and bottom) objectives to image the sample. This has the added benefit of collecting nearly all the emitted fluorescence from the sample, making it a particularly photon-efficient technique.
Signal from both objectives is recombined and allowed to interfere via a custom 3-way beam splitter. The beamsplitter is engineered such that it produces interference images on three synchronized cameras simultaneously. The relative single molecule fluorescence intensities seen by each camera depend strongly on the path length difference taken by the fluorescent photon through both objectives. Thus, fluorophores located closer to the “top” objective will display different relative intensities than those located closer to the “bottom” objective. By carefully calibrating a model fluorophore position against the image intensities in each camera, its precise z-position can be found. Figure 2 illustrates the basic setup for z-localization in iPALM. Note that this three-path setup (rather than the conventional two-path geometry of conventional interferometry) gives two critical advantages. Most importantly, because z-position is calculated based on relative intensities across channels, a two-path detection scheme does not offer a unique intensity ratio for any given z-position within the detection volume. Secondly, at z-positions with relatively high/low intensity ratios, noise can negatively impact the accuracy of the z-position determination.
The iPALM can offer electron microscopy level resolution in intact fluorescently labeled samples. However, as in all SML super-resolution approaches, the achievable resolution depends critically on the number of detected photons, as well as background level. Increasing the number of detected photons from the single emitter and decreasing background signal will favorably affect the overall resolution of the final image. In thin biological samples such as cell culture monolayers, axial and lateral resolutions of <25nm have been demonstrated.
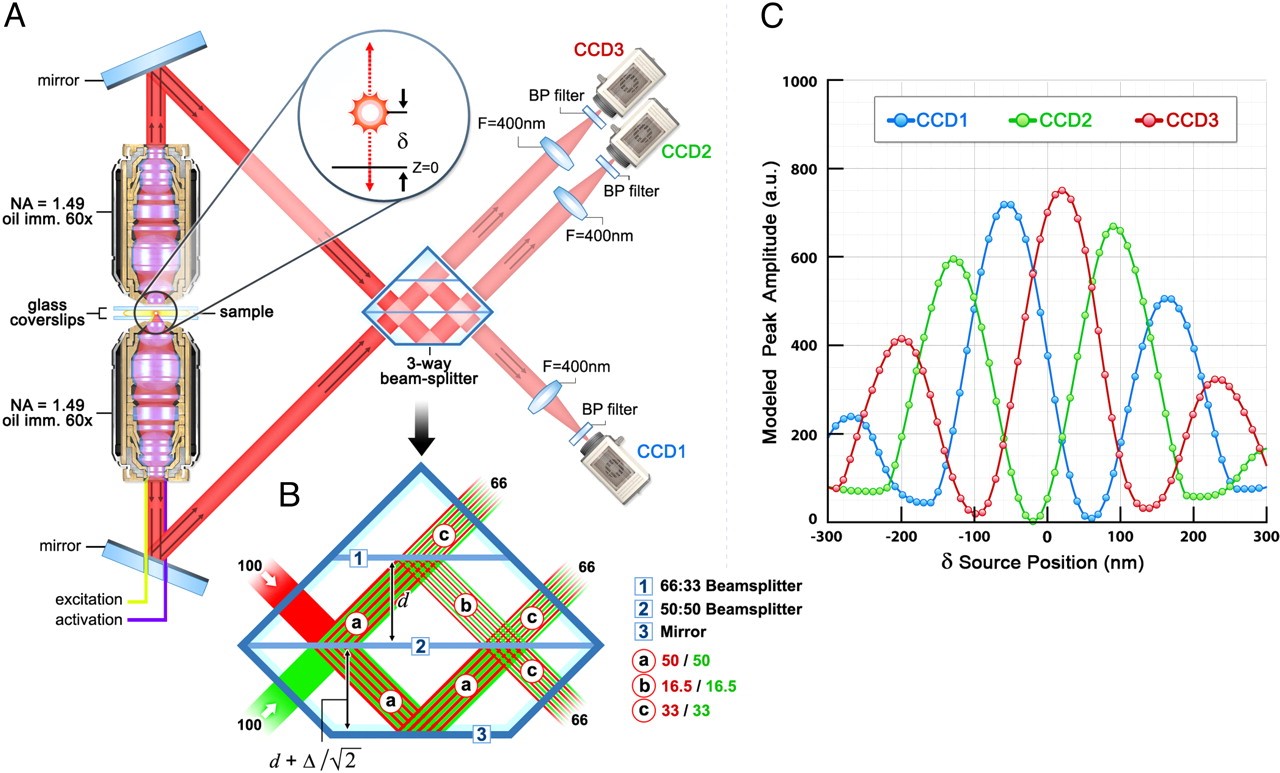
Figure 2. iPALM setup. From Shtengel et al., PNAS (2009)
Specifications
Objectives
- Dual Nikon 60X NA 1.49
Available excitation/activation wavelengths
- 488 nm (~400 W/cm2), 561 nm (~1 kW/cm2), and 640 nm (~3 kW/cm2), excitation lasers
- 405 nm ( ~2-10 W/cm2) laser for photoactivation
Emission filters
- FF01-520/35 (Semrock), FF01-593/40 (Semrock), LP02-647RU (Semrock), FF01-720/SP (Semrock), HQ795/50m (Semrock)
Cameras
- 3 iXon3-DU897E EMCCD (Andor Technologies)
iPALM Limitations
- Temporal resolution is limited due to the high number (~103-403) raw acquisitions need to create SR image. To image dynamic processes, compromises are needed in either sampling density or number of photons to be detected.
- Higher excitation intensity (ca 103 W/cm2) needed than SIM (101-102 W/cm2), thus limiting with highly photon-sensitive samples
- Specialized sample preparation needed. This includes either (1) photoswitchable/photoactivatable proteins and/or (2) certain organic dyes in combination with sample buffer formulations to inhibit irreversible photobleaching.
- Because the iPALM uses TIRF (or near-TIRF) illumination, it is limited to imaging single cells, with <750nm in total imaging depth above the coverslip (see Brown TA, et al., PNAS 2011).
- Importantly, gaining reliable quantitative information relies in large part to the efficiency of sample labeling. Resolution will in part be determined by the extent to which biological structures are faithfully represented by their associated fluorophores. Thus, samples which are “under-labeled” may have poorer apparent resolution. However, samples that are “over-labeled” may be difficult to image due to an inability to render a sufficient number of fluorophores in the dark state to ensure they are isolated both spatially and temporally.
iPALM Compatible Fluorescent Probes
The following table represents a collection of some of the most common iPALM-compatible probes. It is by no means comprehensive. Potential users are urged to read the articles by Dempsey and Shcherbakova, (see Suggested Reading) for a more in-depth discussion, and to contact AIC staff members before submitting their proposals.
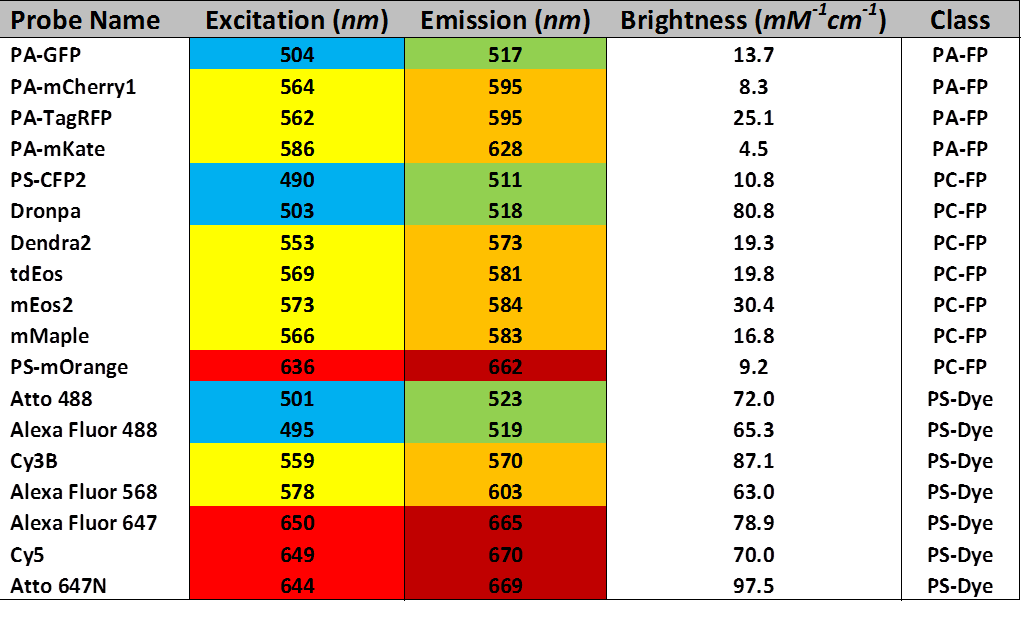
Notes:
Color shading indicates approximate excitation/emission wavelengths. Probes with the same color scheme will use the same laser and filter configurations, and thus cannot be used together in multi-color experiments.
Brightness is calculated as the product of each probe’s extinction coefficient and quantum yield.
PA-FP refers to photo-activatable fluorescent protein; PC-FP refers to photo-convertible fluorescent protein; and PS-Dye refers to any organic small molecule fluorophore that undergoes reversible photoswitching.
Suggested Reading
⦁ Betzig, Eric, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313.5793 (2006): 1642-1645.
⦁ Patterson, George, et al. Superresolution imaging using single-molecule localization. Annual review of physical chemistry 61 (2010): 345.
⦁ Allen, John R., Stephen T. Ross, and Michael W. Davidson. Sample preparation for single molecule localization microscopy. Physical Chemistry Chemical Physics 15.43 (2013): 18771-18783.
⦁ Endesfelder, Ulrike, and Mike Heilemann. Art and artifacts in single-molecule localization microscopy: beyond attractive images. Nature methods 11.3 (2014): 235-238.
⦁ Small, Alex, and Shane Stahlheber. Fluorophore localization algorithms for super-resolution microscopy. Nature methods 11.3 (2014): 267-279.
⦁ Shtengel, Gleb, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proceedings of the National Academy of Sciences 106.9 (2009): 3125-3130.
⦁ Kanchanawong, Pakorn, et al. Nanoscale architecture of integrin-based cell adhesions. Nature 468.7323 (2010): 580-584.
⦁ Kopek, Benjamin G., et al. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proceedings of the National Academy of Sciences 109.16 (2012): 6136-6141.
⦁ Brown TA, Tkachuk A, Shtengel G, et al. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Proceedings of the National Academy of Sciences 31. 24 (2011): 4994-5010.
⦁Dempsey GT, et al. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging Nat. Meth. 8(12) (2011):1027-1036
⦁ Shcherbakova, DM, et al. Photocontrollable Fluorescent Proteins for Superresolution Imaging. Annu. Rev. Biophys. 43 (2014):303-29