Filter
Associated Lab
- Ahrens Lab (2) Apply Ahrens Lab filter
- Betzig Lab (9) Apply Betzig Lab filter
- Hess Lab (1) Apply Hess Lab filter
- Ji Lab (2) Apply Ji Lab filter
- Lavis Lab (1) Apply Lavis Lab filter
- Pedram Lab (2) Apply Pedram Lab filter
- Saalfeld Lab (1) Apply Saalfeld Lab filter
- Remove Shroff Lab filter Shroff Lab
- Wang (Shaohe) Lab (2) Apply Wang (Shaohe) Lab filter
Associated Project Team
Publication Date
Type of Publication
31 Publications
Showing 11-20 of 31 results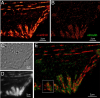
Accurate determination of the relative positions of proteins within localized regions of the cell is essential for understanding their biological function. Although fluorescent fusion proteins are targeted with molecular precision, the position of these genetically expressed reporters is usually known only to the resolution of conventional optics ( approximately 200 nm). Here, we report the use of two-color photoactivated localization microscopy (PALM) to determine the ultrastructural relationship between different proteins fused to spectrally distinct photoactivatable fluorescent proteins (PA-FPs). The nonperturbative incorporation of these endogenous tags facilitates an imaging resolution in whole, fixed cells of approximately 20-30 nm at acquisition times of 5-30 min. We apply the technique to image different pairs of proteins assembled in adhesion complexes, the central attachment points between the cytoskeleton and the substrate in migrating cells. For several pairs, we find that proteins that seem colocalized when viewed by conventional optics are resolved as distinct interlocking nano-aggregates when imaged via PALM. The simplicity, minimal invasiveness, resolution, and speed of the technique all suggest its potential to directly visualize molecular interactions within cellular structures at the nanometer scale.
Commentary: Identifies the photoactivatable fluorescent proteins (PA-FPs) Dronpa and PS-CFP2 as green partners to orange-red PA-FPs such as Kaede and Eos for dual color PALM imaging. Very low crosstalk is demonstrated between the two color channels. Furthermore, since the probes are genetically expressed, they are closely bound to their target proteins and exhibit zero non-specific background. All these properties are essential to unambiguously identify regions of co-localization or separate compartmentalization at the nanoscale, as demonstrated in the examples here.
Fluorescence microscopy is essential for biological research, offering high-contrast imaging of microscopic structures. However, the quality of these images is often compromised by optical aberrations and noise, particularly in low signal-to-noise ratio (SNR) conditions. While adaptive optics (AO) can correct aberrations, it requires costly hardware and slows down imaging; whereas current denoising approaches boost the SNR but leave out the aberration compensation. To address these limitations, we introduce HD2Net, a deep learning framework that enhances image quality by simultaneously denoising and suppressing the effect of aberrations without the need for additional hardware. Building on our previous work, HD2Net incorporates noise estimation and aberration removal modules, effectively restoring images degraded by noise and aberrations. Through comprehensive evaluation of synthetic phantoms and biological data, we demonstrate that HD2Net outperforms existing methods, significantly improving image resolution and contrast. This framework offers a promising solution for enhancing biological imaging, particularly in challenging aberrating and low-light conditions.
Fluorescence microscopy is essential for biological research, offering high-contrast imaging of microscopic structures. However, the quality of these images is often compromised by optical aberrations and noise, particularly in low signal-to-noise ratio (SNR) conditions. While adaptive optics (AO) can correct aberrations, it requires costly hardware and slows down imaging; whereas current denoising approaches boost the SNR but leave out the aberration compensation. To address these limitations, we introduce HD2Net, a deep learning framework that enhances image quality by simultaneously denoising and suppressing the effect of aberrations without the need for additional hardware. Building on our previous work, HD2Net incorporates noise estimation and aberration removal modules, effectively restoring images degraded by noise and aberrations. Through comprehensive evaluation of synthetic phantoms and biological data, we demonstrate that HD2Net outperforms existing methods, significantly improving image resolution and contrast. This framework offers a promising solution for enhancing biological imaging, particularly in challenging aberrating and low-light conditions.
We combined photoactivated localization microscopy (PALM) with live-cell single-particle tracking to create a new method termed sptPALM. We created spatially resolved maps of single-molecule motions by imaging the membrane proteins Gag and VSVG, and obtained several orders of magnitude more trajectories per cell than traditional single-particle tracking enables. By probing distinct subsets of molecules, sptPALM can provide insight into the origins of spatial and temporal heterogeneities in membranes.
Commentary: As a stepping stone to true live cell PALM (see above), our collaborator Jennifer Lippincott-Schwartz suggested using the sparse photoactivation principle of PALM to track the nanoscale motion of thousands of individual molecules within a single living cell. Termed single particle tracking PALM (sptPALM), Jennifer’s postdocs Suliana Manley and Jen Gillette used the method in our PALM rig to create spatially resolved maps of diffusion rates in the plasma membrane of live cells. sptPALM is a powerful tool to study the active cytoskeletal or passive diffusional transport of individual molecules with far more measurements per cell than is possible without sparse photoactivation.
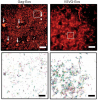
All multicellular systems produce and dynamically regulate extracellular matrices (ECM) that play important roles in both biochemical and mechanical signaling. Though the spatial arrangement of these extracellular assemblies is critical to their biological functions, visualization of ECM structure is challenging, in part because the biomolecules that compose the ECM are difficult to fluorescently label individually and collectively. Here, we present a cell-impermeable small molecule fluorophore, termed Rhobo6, that turns on and red shifts upon reversible binding to glycans. Given that most ECM components are densely glycosylated, the dye enables wash-free visualization of ECM, in systems ranging from in vitro substrates to in vivo mouse mammary tumors. Relative to existing techniques, Rhobo6 provides a broad substrate profile, superior tissue penetration, nonperturbative labeling, and negligible photobleaching. This work establishes a straightforward method for imaging the distribution of ECM in live tissues and organisms, lowering barriers for investigation of extracellular biology.
All multicellular systems produce and dynamically regulate extracellular matrices (ECMs) that play essential roles in both biochemical and mechanical signaling. Though the spatial arrangement of these extracellular assemblies is critical to their biological functions, visualization of ECM structure is challenging, in part because the biomolecules that compose the ECM are difficult to fluorescently label individually and collectively. Here, we present a cell-impermeable small-molecule fluorophore, termed Rhobo6, that turns on and red shifts upon reversible binding to glycans. Given that most ECM components are densely glycosylated, the dye enables wash-free visualization of ECM, in systems ranging from in vitro substrates to in vivo mouse mammary tumors. Relative to existing techniques, Rhobo6 provides a broad substrate profile, superior tissue penetration, non-perturbative labeling, and negligible photobleaching. This work establishes a straightforward method for imaging the distribution of ECM in live tissues and organisms, lowering barriers for investigation of extracellular biology.
The proliferation of microscopy methods for live-cell imaging offers many new possibilities for users but can also be challenging to navigate. The prevailing challenge in live-cell fluorescence microscopy is capturing intra-cellular dynamics while preserving cell viability. Computational methods can help to address this challenge and are now shifting the boundaries of what is possible to capture in living systems. In this Review, we discuss these computational methods focusing on artificial intelligence-based approaches that can be layered on top of commonly used existing microscopies as well as hybrid methods that integrate computation and microscope hardware. We specifically discuss how computational approaches can improve the signal-to-noise ratio, spatial resolution, temporal resolution and multi-colour capacity of live-cell imaging.
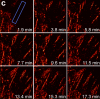
We demonstrate live-cell super-resolution imaging using photoactivated localization microscopy (PALM). The use of photon-tolerant cell lines in combination with the high resolution and molecular sensitivity of PALM permitted us to investigate the nanoscale dynamics within individual adhesion complexes (ACs) in living cells under physiological conditions for as long as 25 min, with half of the time spent collecting the PALM images at spatial resolutions down to approximately 60 nm and frame rates as short as 25 s. We visualized the formation of ACs and measured the fractional gain and loss of individual paxillin molecules as each AC evolved. By allowing observation of a wide variety of nanoscale dynamics, live-cell PALM provides insights into molecular assembly during the initiation, maturation and dissolution of cellular processes.
Commentary: The first example of true live cell and time lapse imaging by localization microscopy (as opposed to particle tracking), this paper uses the Nyquist criterion to establish a necessary condition for true spatial resolution based on the density of localized molecules – a condition often unmet in claims elsewhere in the superresolution literature.
By any method, higher spatiotemporal resolution requires increasing light exposure at the specimen, making noninvasive imaging increasingly difficult. Here, simultaneous differential interference contrast imaging is used to establish that cells behave physiologically before, during, and after PALM imaging. Similar controls are lacking from many supposed “live cell” superresolution demonstrations.
Stereocilia are unidirectional F-actin-based cylindrical protrusions on the apical surface of inner ear hair cells and function as biological mechanosensors of sound and acceleration. Development of functional stereocilia requires motor activities of unconventional myosins to transport proteins necessary for elongating the F-actin cores and to assemble the mechanoelectrical transduction (MET) channel complex. However, how each myosin localizes in stereocilia using the energy from ATP hydrolysis is only partially understood. In this study, we develop a methodology for live-cell single-molecule fluorescence microscopy of organelles protruding from the apical surface using a dual-view light-sheet microscope, diSPIM. We demonstrate that MYO7A, a component of the MET machinery, traffics as a dimer in stereocilia. Movements of MYO7A are restricted when scaffolded by the plasma membrane and F-actin as mediated by MYO7A’s interacting partners. Here, we discuss the technical details of our methodology and its future applications including analyses of cargo transportation in various organelles.
Spatial information is critical to the interrogation of developmental and tissue-level regulation of gene expression. However, this information is usually lost when global mRNA levels from tissues are measured using reverse transcriptase PCR, microarray analysis or high-throughput sequencing. By contrast, single-molecule fluorescence in situ hybridization (smFISH) preserves the spatial information of the cellular mRNA content with subcellular resolution within tissues. Here we describe an smFISH protocol that allows for the quantification of single mRNAs in Drosophila embryos, using commercially available smFISH probes (e.g., short fluorescently labeled DNA oligonucleotides) in combination with wide-field epifluorescence, confocal or instant structured illumination microscopy (iSIM, a super-resolution imaging approach) and a spot-detection algorithm. Fixed Drosophila embryos are hybridized in solution with a mixture of smFISH probes, mounted onto coverslips and imaged in 3D. Individual fluorescently labeled mRNAs are then localized within tissues and counted using spot-detection software to generate quantitative, spatially resolved gene expression data sets. With minimum guidance, a graduate student can successfully implement this protocol. The smFISH procedure described here can be completed in 4-5 d.