Filter
Associated Lab
- Aso Lab (1) Apply Aso Lab filter
- Remove Betzig Lab filter Betzig Lab
- Bock Lab (1) Apply Bock Lab filter
- Clapham Lab (2) Apply Clapham Lab filter
- Fetter Lab (2) Apply Fetter Lab filter
- Harris Lab (7) Apply Harris Lab filter
- Hess Lab (8) Apply Hess Lab filter
- Ji Lab (11) Apply Ji Lab filter
- Lavis Lab (8) Apply Lavis Lab filter
- Lippincott-Schwartz Lab (6) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (7) Apply Liu (Zhe) Lab filter
- Magee Lab (2) Apply Magee Lab filter
- Rubin Lab (1) Apply Rubin Lab filter
- Saalfeld Lab (2) Apply Saalfeld Lab filter
- Schreiter Lab (1) Apply Schreiter Lab filter
- Shroff Lab (9) Apply Shroff Lab filter
- Singer Lab (1) Apply Singer Lab filter
- Svoboda Lab (2) Apply Svoboda Lab filter
- Tjian Lab (4) Apply Tjian Lab filter
- Turner Lab (1) Apply Turner Lab filter
Associated Project Team
Publication Date
- 2025 (2) Apply 2025 filter
- 2024 (2) Apply 2024 filter
- 2023 (4) Apply 2023 filter
- 2022 (3) Apply 2022 filter
- 2021 (2) Apply 2021 filter
- 2020 (4) Apply 2020 filter
- 2019 (7) Apply 2019 filter
- 2018 (6) Apply 2018 filter
- 2017 (8) Apply 2017 filter
- 2016 (12) Apply 2016 filter
- 2015 (11) Apply 2015 filter
- 2014 (8) Apply 2014 filter
- 2013 (4) Apply 2013 filter
- 2012 (5) Apply 2012 filter
- 2011 (7) Apply 2011 filter
- 2010 (3) Apply 2010 filter
- 2009 (2) Apply 2009 filter
- 2008 (8) Apply 2008 filter
- 2007 (2) Apply 2007 filter
- 2006 (1) Apply 2006 filter
- 2005 (1) Apply 2005 filter
- 1995 (1) Apply 1995 filter
- 1994 (2) Apply 1994 filter
- 1993 (2) Apply 1993 filter
- 1992 (4) Apply 1992 filter
- 1991 (2) Apply 1991 filter
Type of Publication
113 Publications
Showing 91-100 of 113 resultsRecent advances in optical microscopy have enabled biological imaging beyond the diffraction limit at nanometer resolution. A general feature of most of the techniques based on photoactivated localization microscopy (PALM) or stochastic optical reconstruction microscopy (STORM) has been the use of thin biological samples in combination with total internal reflection, thus limiting the imaging depth to a fraction of an optical wavelength. However, to study whole cells or organelles that are typically up to 15 microm deep into the cell, the extension of these methods to a three-dimensional (3D) super resolution technique is required. Here, we report an advance in optical microscopy that enables imaging of protein distributions in cells with a lateral localization precision better than 50 nm at multiple imaging planes deep in biological samples. The approach is based on combining the lateral super resolution provided by PALM with two-photon temporal focusing that provides optical sectioning. We have generated super-resolution images over an axial range of approximately 10 microm in both mitochondrially labeled fixed cells, and in the membranes of living S2 Drosophila cells.
Neurobiological processes occur on spatiotemporal scales spanning many orders of magnitude. Greater understanding of these processes therefore demands improvements in the tools used in their study. Here we review recent efforts to enhance the speed and resolution of one such tool, fluorescence microscopy, with an eye toward its application to neurobiological problems. On the speed front, improvements in beam scanning technology, signal generation rates, and photodamage mediation are bringing us closer to the goal of real-time functional imaging of extended neural networks. With regard to resolution, emerging methods of adaptive optics may lead to diffraction-limited imaging or much deeper imaging in optically inhomogeneous tissues, and super-resolution techniques may prove a powerful adjunct to electron microscopic methods for nanometric neural circuit reconstruction.
Neurobiological processes occur on spatiotemporal scales spanning many orders of magnitude. Greater understanding of these processes therefore demands improvements in the tools used in their study. Here we review recent efforts to enhance the speed and resolution of one such tool, fluorescence microscopy, with an eye toward its application to neurobiological problems. On the speed front, improvements in beam scanning technology, signal generation rates, and photodamage mediation are bringing us closer to the goal of real-time functional imaging of extended neural networks. With regard to resolution, emerging methods of adaptive optics may lead to diffraction-limited imaging or much deeper imaging in optically inhomogeneous tissues, and super-resolution techniques may prove a powerful adjunct to electron microscopic methods for nanometric neural circuit reconstruction.
Commentary: A brief review of recent trends in microscopy. The section “Caveats regarding the application of superresolution microscopy” was written in an effort to inject a dose of reality and caution into the unquestioning enthusiasm in the academic community for all things superresolution, covering the topics of labeling density and specificity, sample preparation artifacts, speed vs. resolution vs. photodamage, and the implications of signal-to-background for Nyquist vs. Rayleigh definitions of resolution.

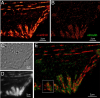
Key to understanding a protein’s biological function is the accurate determination of its spatial distribution inside a cell. Although fluorescent protein markers allow the targeting of specific proteins with molecular precision, much of this information is lost when the resultant fusion proteins are imaged with conventional, diffraction-limited optics. In response, several imaging modalities that are capable of resolution below the diffraction limit (approximately 200 nm) have emerged. Here, both single- and dual-color superresolution imaging of biological structures using photoactivated localization microscopy (PALM) are described. The examples discussed focus on adhesion complexes: dense, protein-filled assemblies that form at the interface between cells and their substrata. A particular emphasis is placed on the instrumentation and photoactivatable fluorescent protein (PA-FP) tags necessary to achieve PALM images at approximately 20 nm resolution in 5 to 30 min in fixed cells.
Commentary: A paper spearheaded by Hari which gives a thorough description of the methods and hardware needed to successfully practice PALM, including cover slip preparation, cell transfection and fixation, drift correction with fiducials, characterization of on/off contrast ratios for different photoactivted fluorescent proteins, identifying PALM-suitable cells, and mechanical and optical components of a PALM system.
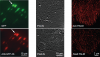
We demonstrate live-cell super-resolution imaging using photoactivated localization microscopy (PALM). The use of photon-tolerant cell lines in combination with the high resolution and molecular sensitivity of PALM permitted us to investigate the nanoscale dynamics within individual adhesion complexes (ACs) in living cells under physiological conditions for as long as 25 min, with half of the time spent collecting the PALM images at spatial resolutions down to approximately 60 nm and frame rates as short as 25 s. We visualized the formation of ACs and measured the fractional gain and loss of individual paxillin molecules as each AC evolved. By allowing observation of a wide variety of nanoscale dynamics, live-cell PALM provides insights into molecular assembly during the initiation, maturation and dissolution of cellular processes.
Commentary: The first example of true live cell and time lapse imaging by localization microscopy (as opposed to particle tracking), this paper uses the Nyquist criterion to establish a necessary condition for true spatial resolution based on the density of localized molecules – a condition often unmet in claims elsewhere in the superresolution literature.
By any method, higher spatiotemporal resolution requires increasing light exposure at the specimen, making noninvasive imaging increasingly difficult. Here, simultaneous differential interference contrast imaging is used to establish that cells behave physiologically before, during, and after PALM imaging. Similar controls are lacking from many supposed “live cell” superresolution demonstrations.
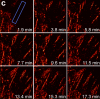
We combined photoactivated localization microscopy (PALM) with live-cell single-particle tracking to create a new method termed sptPALM. We created spatially resolved maps of single-molecule motions by imaging the membrane proteins Gag and VSVG, and obtained several orders of magnitude more trajectories per cell than traditional single-particle tracking enables. By probing distinct subsets of molecules, sptPALM can provide insight into the origins of spatial and temporal heterogeneities in membranes.
Commentary: As a stepping stone to true live cell PALM (see above), our collaborator Jennifer Lippincott-Schwartz suggested using the sparse photoactivation principle of PALM to track the nanoscale motion of thousands of individual molecules within a single living cell. Termed single particle tracking PALM (sptPALM), Jennifer’s postdocs Suliana Manley and Jen Gillette used the method in our PALM rig to create spatially resolved maps of diffusion rates in the plasma membrane of live cells. sptPALM is a powerful tool to study the active cytoskeletal or passive diffusional transport of individual molecules with far more measurements per cell than is possible without sparse photoactivation.
Pulsed lasers are key elements in nonlinear bioimaging techniques such as two-photon fluorescence excitation (TPE) microscopy. Typically, however, only a percent or less of the laser power available can be delivered to the sample before photoinduced damage becomes excessive. Here we describe a passive pulse splitter that converts each laser pulse into a fixed number of sub-pulses of equal energy. We applied the splitter to TPE imaging of fixed mouse brain slices labeled with GFP and show that, in different power regimes, the splitter can be used either to increase the signal rate more than 100-fold or to reduce the rate of photobleaching by over fourfold. In living specimens, the gains were even greater: a ninefold reduction in photobleaching during in vivo imaging of Caenorhabditis elegans larvae, and a six- to 20-fold decrease in the rate of photodamage during calcium imaging of rat hippocampal brain slices.

Pulsed lasers are key elements in nonlinear bioimaging techniques such as two-photon fluorescence excitation (TPE) microscopy. Typically, however, only a percent or less of the laser power available can be delivered to the sample before photoinduced damage becomes excessive. Here we describe a passive pulse splitter that converts each laser pulse into a fixed number of sub-pulses of equal energy. We applied the splitter to TPE imaging of fixed mouse brain slices labeled with GFP and show that, in different power regimes, the splitter can be used either to increase the signal rate more than 100-fold or to reduce the rate of photobleaching by over fourfold. In living specimens, the gains were even greater: a ninefold reduction in photobleaching during in vivo imaging of Caenorhabditis elegans larvae, and a six- to 20-fold decrease in the rate of photodamage during calcium imaging of rat hippocampal brain slices.
Commentary: Na Ji came to me early in her postdoc with an idea to reduce photodamage in nonlinear microscopy by splitting the pulses from an ultrafast laser into multiple subpulses of reduced energy. In six weeks, we constructed a prototype pulse splitter and obtained initial results confirming the validity of her vision. Further experiments with Jeff Magee demonstrated that the splitter could be used to increase imaging speed or reduce photodamage in two photon microscopy by one to two orders of magnitude. This project is a great example of how quickly one can react and exploit new ideas in the Janelia environment.
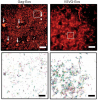
Accurate determination of the relative positions of proteins within localized regions of the cell is essential for understanding their biological function. Although fluorescent fusion proteins are targeted with molecular precision, the position of these genetically expressed reporters is usually known only to the resolution of conventional optics ( approximately 200 nm). Here, we report the use of two-color photoactivated localization microscopy (PALM) to determine the ultrastructural relationship between different proteins fused to spectrally distinct photoactivatable fluorescent proteins (PA-FPs). The nonperturbative incorporation of these endogenous tags facilitates an imaging resolution in whole, fixed cells of approximately 20-30 nm at acquisition times of 5-30 min. We apply the technique to image different pairs of proteins assembled in adhesion complexes, the central attachment points between the cytoskeleton and the substrate in migrating cells. For several pairs, we find that proteins that seem colocalized when viewed by conventional optics are resolved as distinct interlocking nano-aggregates when imaged via PALM. The simplicity, minimal invasiveness, resolution, and speed of the technique all suggest its potential to directly visualize molecular interactions within cellular structures at the nanometer scale.
Commentary: Identifies the photoactivatable fluorescent proteins (PA-FPs) Dronpa and PS-CFP2 as green partners to orange-red PA-FPs such as Kaede and Eos for dual color PALM imaging. Very low crosstalk is demonstrated between the two color channels. Furthermore, since the probes are genetically expressed, they are closely bound to their target proteins and exhibit zero non-specific background. All these properties are essential to unambiguously identify regions of co-localization or separate compartmentalization at the nanoscale, as demonstrated in the examples here.
In conventional biological imaging, diffraction places a limit on the minimal xy distance at which two marked objects can be discerned. Consequently, resolution of target molecules within cells is typically coarser by two orders of magnitude than the molecular scale at which the proteins are spatially distributed. Photoactivated localization microscopy (PALM) optically resolves selected subsets of protect fluorescent probes within cells at mean separations of <25 nanometers. It involves serial photoactivation and subsequent photobleaching of numerous sparse subsets of photoactivated fluorescent protein molecules. Individual molecules are localized at near molecular resolution by determining their centers of fluorescent emission via a statistical fit of their point-spread-function. The position information from all subsets is then assembled into a super-resolution image, in which individual fluorescent molecules are isolated at high molecular densities. In this paper, some of the limitations for PALM imaging under current experimental conditions are discussed.