Filter
Associated Lab
- Ahrens Lab (3) Apply Ahrens Lab filter
- Aso Lab (3) Apply Aso Lab filter
- Baker Lab (5) Apply Baker Lab filter
- Betzig Lab (8) Apply Betzig Lab filter
- Branson Lab (6) Apply Branson Lab filter
- Card Lab (1) Apply Card Lab filter
- Cardona Lab (1) Apply Cardona Lab filter
- Chklovskii Lab (2) Apply Chklovskii Lab filter
- Cui Lab (2) Apply Cui Lab filter
- Darshan Lab (1) Apply Darshan Lab filter
- Dickson Lab (6) Apply Dickson Lab filter
- Druckmann Lab (1) Apply Druckmann Lab filter
- Dudman Lab (4) Apply Dudman Lab filter
- Eddy/Rivas Lab (3) Apply Eddy/Rivas Lab filter
- Egnor Lab (1) Apply Egnor Lab filter
- Fetter Lab (1) Apply Fetter Lab filter
- Fitzgerald Lab (1) Apply Fitzgerald Lab filter
- Freeman Lab (3) Apply Freeman Lab filter
- Gonen Lab (10) Apply Gonen Lab filter
- Grigorieff Lab (3) Apply Grigorieff Lab filter
- Harris Lab (2) Apply Harris Lab filter
- Heberlein Lab (2) Apply Heberlein Lab filter
- Hermundstad Lab (2) Apply Hermundstad Lab filter
- Hess Lab (5) Apply Hess Lab filter
- Jayaraman Lab (1) Apply Jayaraman Lab filter
- Ji Lab (3) Apply Ji Lab filter
- Johnson Lab (1) Apply Johnson Lab filter
- Kainmueller Lab (2) Apply Kainmueller Lab filter
- Karpova Lab (1) Apply Karpova Lab filter
- Keller Lab (8) Apply Keller Lab filter
- Lavis Lab (7) Apply Lavis Lab filter
- Lee (Albert) Lab (4) Apply Lee (Albert) Lab filter
- Leonardo Lab (4) Apply Leonardo Lab filter
- Li Lab (1) Apply Li Lab filter
- Lippincott-Schwartz Lab (12) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (4) Apply Liu (Zhe) Lab filter
- Looger Lab (11) Apply Looger Lab filter
- Magee Lab (1) Apply Magee Lab filter
- Menon Lab (3) Apply Menon Lab filter
- Murphy Lab (1) Apply Murphy Lab filter
- Pavlopoulos Lab (2) Apply Pavlopoulos Lab filter
- Reiser Lab (2) Apply Reiser Lab filter
- Riddiford Lab (4) Apply Riddiford Lab filter
- Romani Lab (2) Apply Romani Lab filter
- Rubin Lab (9) Apply Rubin Lab filter
- Saalfeld Lab (1) Apply Saalfeld Lab filter
- Scheffer Lab (7) Apply Scheffer Lab filter
- Sgro Lab (1) Apply Sgro Lab filter
- Simpson Lab (2) Apply Simpson Lab filter
- Singer Lab (10) Apply Singer Lab filter
- Spruston Lab (1) Apply Spruston Lab filter
- Stern Lab (6) Apply Stern Lab filter
- Sternson Lab (5) Apply Sternson Lab filter
- Stringer Lab (1) Apply Stringer Lab filter
- Svoboda Lab (7) Apply Svoboda Lab filter
- Tebo Lab (2) Apply Tebo Lab filter
- Tervo Lab (1) Apply Tervo Lab filter
- Tillberg Lab (1) Apply Tillberg Lab filter
- Tjian Lab (4) Apply Tjian Lab filter
- Truman Lab (1) Apply Truman Lab filter
- Turaga Lab (1) Apply Turaga Lab filter
- Turner Lab (1) Apply Turner Lab filter
- Wang (Shaohe) Lab (1) Apply Wang (Shaohe) Lab filter
- Wu Lab (2) Apply Wu Lab filter
- Zlatic Lab (1) Apply Zlatic Lab filter
- Zuker Lab (1) Apply Zuker Lab filter
Associated Project Team
Publication Date
- December 2014 (35) Apply December 2014 filter
- November 2014 (14) Apply November 2014 filter
- October 2014 (15) Apply October 2014 filter
- September 2014 (17) Apply September 2014 filter
- August 2014 (14) Apply August 2014 filter
- July 2014 (26) Apply July 2014 filter
- June 2014 (14) Apply June 2014 filter
- May 2014 (14) Apply May 2014 filter
- April 2014 (20) Apply April 2014 filter
- March 2014 (18) Apply March 2014 filter
- February 2014 (15) Apply February 2014 filter
- January 2014 (34) Apply January 2014 filter
- Remove 2014 filter 2014
Type of Publication
236 Publications
Showing 21-30 of 236 resultsThe self-assembly of proteins into highly ordered nanoscale architectures is a hallmark of biological systems. The sophisticated functions of these molecular machines have inspired the development of methods to engineer self-assembling protein nanostructures; however, the design of multi-component protein nanomaterials with high accuracy remains an outstanding challenge. Here we report a computational method for designing protein nanomaterials in which multiple copies of two distinct subunits co-assemble into a specific architecture. We use the method to design five 24-subunit cage-like protein nanomaterials in two distinct symmetric architectures and experimentally demonstrate that their structures are in close agreement with the computational design models. The accuracy of the method and the number and variety of two-component materials that it makes accessible suggest a route to the construction of functional protein nanomaterials tailored to specific applications.
In this work we present a novel technique we term active graph matching, which integrates the popular active shape model into a sparse graph matching problem. This way we are able to combine the benefits of a global, statistical deformation model with the benefits of a local deformation model in form of a second-order random field. We present a new iterative energy minimization technique which achieves empirically good results. This enables us to exceed state-of-the art results for the task of annotating nuclei in 3D microscopic images of C. elegans. Furthermore with the help of the generalized Hough transform we are able to jointly segment and annotate a large set of nuclei in a fully automatic fashion for the first time.
Rod photoreceptors contribute to vision over an ∼ 6-log-unit range of light intensities. The wide dynamic range of rod vision is thought to depend upon light intensity-dependent switching between two parallel pathways linking rods to ganglion cells: a rod → rod bipolar (RB) cell pathway that operates at dim backgrounds and a rod → cone → cone bipolar cell pathway that operates at brighter backgrounds. We evaluated this conventional model of rod vision by recording rod-mediated light responses from ganglion and AII amacrine cells and by recording RB-mediated synaptic currents from AII amacrine cells in mouse retina. Contrary to the conventional model, we found that the RB pathway functioned at backgrounds sufficient to activate the rod → cone pathway. As background light intensity increased, the RB's role changed from encoding the absorption of single photons to encoding contrast modulations around mean luminance. This transition is explained by the intrinsic dynamics of transmission from RB synapses.
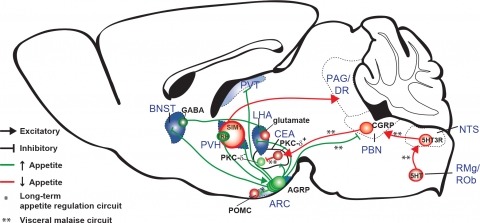
New tools for mapping and manipulating molecularly defined neural circuits have improved understanding of how the central nervous system regulates appetite. Studies focused on AGRP neurons, a starvation-sensitive hypothalamic population, have identified multiple circuit elements that can elicit or suppress feeding behavior. Distinct axon projections of this neuron population point to different circuits that regulate long-term appetite, short-term feeding, or visceral malaise-mediated anorexia. Here, we review recent studies examining these neural circuits that control food intake. © 2014 S. Karger AG, Basel.
Recent reports have associated NCF2, encoding a core component of the multi-protein NADPH oxidase (NADPHO), with systemic lupus erythematosus (SLE) susceptibility in individuals of European ancestry. To identify ethnicity-specific and -robust variants within NCF2, we assessed 145 SNPs in and around the NCF2 gene in 5325 cases and 21 866 controls of European-American (EA), African-American (AA), Hispanic (HS) and Korean (KR) ancestry. Subsequent imputation, conditional, haplotype and bioinformatic analyses identified seven potentially functional SLE-predisposing variants. Association with non-synonymous rs17849502, previously reported in EA, was detected in EA, HS and AA (P(EA) = 1.01 × 10(-54), PHS = 3.68 × 10(-10), P(AA) = 0.03); synonymous rs17849501 was similarly significant. These SNPs were monomorphic in KR. Novel associations were detected with coding variants at rs35937854 in AA (PAA = 1.49 × 10(-9)), and rs13306575 in HS and KR (P(HS) = 7.04 × 10(-7), P(KR) = 3.30 × 10(-3)). In KR, a 3-SNP haplotype was significantly associated (P = 4.20 × 10(-7)), implying that SLE predisposing variants were tagged. Significant SNP-SNP interaction (P = 0.02) was detected between rs13306575 and rs17849502 in HS, and a dramatically increased risk (OR = 6.55) with a risk allele at each locus. Molecular modeling predicts that these non-synonymous mutations could disrupt NADPHO complex assembly. The risk allele of rs17849501, located in a conserved transcriptional regulatory region, increased reporter gene activity, suggesting in vivo enhancer function. Our results not only establish allelic heterogeneity within NCF2 associated with SLE, but also emphasize the utility of multi-ethnic cohorts to identify predisposing variants explaining additional phenotypic variance ('missing heritability') of complex diseases like SLE.
For over 50 years, amphotericin has remained the powerful but highly toxic last line of defense in treating life-threatening fungal infections in humans with minimal development of microbial resistance. Understanding how this small molecule kills yeast is thus critical for guiding development of derivatives with an improved therapeutic index and other resistance-refractory antimicrobial agents. In the widely accepted ion channel model for its mechanism of cytocidal action, amphotericin forms aggregates inside lipid bilayers that permeabilize and kill cells. In contrast, we report that amphotericin exists primarily in the form of large, extramembranous aggregates that kill yeast by extracting ergosterol from lipid bilayers. These findings reveal that extraction of a polyfunctional lipid underlies the resistance-refractory antimicrobial action of amphotericin and suggests a roadmap for separating its cytocidal and membrane-permeabilizing activities. This new mechanistic understanding is also guiding development of what are to our knowledge the first derivatives of amphotericin that kill yeast but not human cells.
Intracellular recording allows precise measurement and manipulation of individual neurons, but it requires stable mechanical contact between the electrode and the cell membrane, and thus it has remained challenging to perform in behaving animals. Whole-cell recordings in freely moving animals can be obtained by rigidly fixing ('anchoring') the pipette electrode to the head; however, previous anchoring procedures were slow and often caused substantial pipette movement, resulting in loss of the recording or of recording quality. We describe a UV-transparent collar and UV-cured adhesive technique that rapidly (within 15 s) anchors pipettes in place with virtually no movement, thus substantially improving the reliability, yield and quality of freely moving whole-cell recordings. Recordings are first obtained from anesthetized or awake head-fixed rats. UV light cures the thin adhesive layers linking pipette to collar to head. Then, the animals are rapidly and smoothly released for recording during unrestrained behavior. The anesthetized-patched version can be completed in ∼4-7 h (excluding histology) and the awake-patched version requires ∼1-4 h per day for ∼2 weeks. These advances should greatly facilitate studies of neuronal integration and plasticity in identified cells during natural behaviors.
Many different types of functional non-coding RNAs participate in a wide range of important cellular functions but the large majority of these RNAs are not routinely annotated in published genomes. Several programs have been developed for identifying RNAs, including specific tools tailored to a particular RNA family as well as more general ones designed to work for any family. Many of these tools utilize covariance models (CMs), statistical models of the conserved sequence, and structure of an RNA family. In this chapter, as an illustrative example, the Infernal software package and CMs from the Rfam database are used to identify RNAs in the genome of the archaeon Methanobrevibacter ruminantium, uncovering some additional RNAs not present in the genome’s initial annotation. Analysis of the results and comparison with family-specific methods demonstrate some important strengths and weaknesses of this general approach.
Reconstructing neuronal circuits at the level of synapses is a central problem in neuroscience and becoming a focus of the emerging field of connectomics. To date, electron microscopy (EM) is the most proven technique for identifying and quantifying synaptic connections. As advances in EM make acquiring larger datasets possible, subsequent manual synapse identification ({\em i.e.}, proofreading) for deciphering a connectome becomes a major time bottleneck. Here we introduce a large-scale, high-throughput, and semi-automated methodology to efficiently identify synapses. We successfully applied our methodology to the Drosophila medulla optic lobe, annotating many more synapses than previous connectome efforts. Our approaches are extensible and will make the often complicated process of synapse identification accessible to a wider-community of potential proofreaders.
Aphids evolved novel cells, called bacteriocytes, that differentiate specifically to harbour the obligatory mutualistic endosymbiotic bacteria Buchnera aphidicola. The genome of the host aphid Acyrthosiphon pisum contains many orphan genes that display no similarity with genes found in other sequenced organisms, prompting us to hypothesize that some of these orphan genes are related to lineage-specific traits, such as symbiosis. We conducted deep sequencing of bacteriocytes mRNA followed by whole mount in situ hybridizations of over-represented transcripts encoding aphid-specific orphan proteins. We identified a novel class of genes that encode small proteins with signal peptides, which are often cysteine-rich, that are over-represented in bacteriocytes. These genes are first expressed at a developmental time point coincident with the incorporation of symbionts strictly in the cells that contribute to the bacteriocyte and this bacteriocyte-specific expression is maintained throughout the aphid's life. The expression pattern suggests that recently evolved secretion proteins act within bacteriocytes, perhaps to mediate the symbiosis with beneficial bacterial partners, which is reminiscent of the evolution of novel cysteine-rich secreted proteins of leguminous plants that regulate nitrogen-fixing endosymbionts.