Electron Microscopy of Membrane Channels, Transporters, and Macromolecular Machines
Our research focuses on two fundamental questions in cell biology: How do cells interact with each other and with their environment? How do cells obtain the nutrients essential for their survival? We focus on integral membrane proteins because they play a crucial role in mediating information transfer between cells and between compartments within cells and because they are involved in vital processes such as energy conversion, sensing, fusion, and nutrient and solute uptake. More than 20 percent of the genes in the human genome encode membrane-traversing proteins. Thus it is critical to study structures of membrane proteins to unravel important biological and physiological processes. Our model system is the eye lens, whose cells are rich in junction-forming membrane proteins and transporters needed for nutrient uptake.
Biomembranes represent the interface of life, where homeostasis is controlled and maintained, nutrients and wastes are exchanged, signals are perceived and transmitted, cells are attached and interact, and pathogens break in and invade. My laboratory is interested in revealing the structural and molecular mechanisms by which membrane proteins mediate these cross-membrane processes with high specificity and efficiency. Using high-resolution cryo-electron microscopy (cryo-EM), together with other biophysical and biochemical approaches, we are focusing on membrane channels, transporters, and macromolecular machines that form biological pores.
We use a multidisciplinary approach, including cryo-EM, x-ray crystallography, and nuclear magnetic resonance (NMR) spectroscopy for determining protein structure and characterizing membrane protein complex formation and binding stoichiometries. To assay protein function, we use biophysical techniques with reconstituted proteoliposomes and investigate protein structure under a variety of physiologically important regulatory states.


Membrane Channels: Water Channels
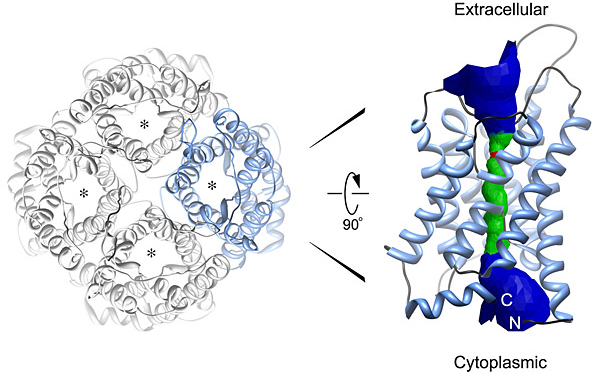
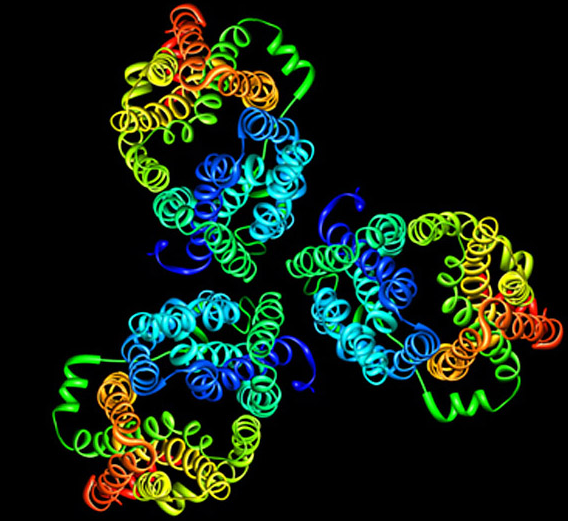
Most of the interior of a cell is water. Life could not exist without circulating water, which removes waste products from cells and solvates nutrients and ions that are vital for our existence. Water can diffuse freely though cell membranes but only at very low rates; for this reason, all three domains of life, from the simplest unicellular organisms to mammals, express aquaporins, membrane proteins that form specialized pores for water. All aquaporins studied to date structurally assemble as tetramers, but each monomer functions independently as a water channel.
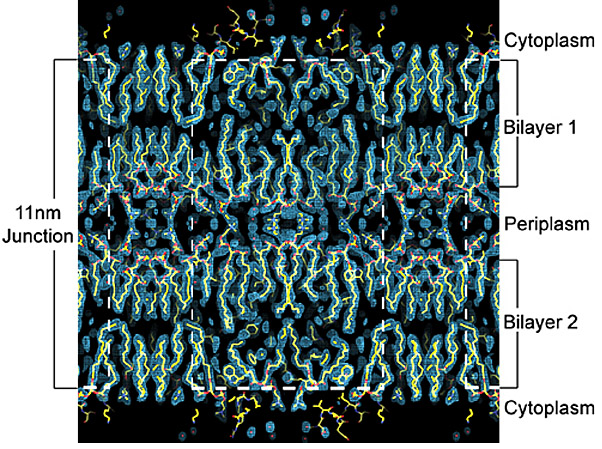
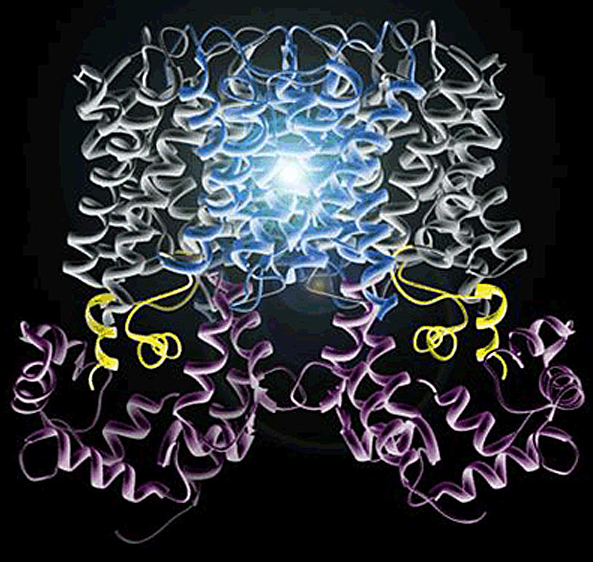
My previous work with Thomas Walz (HHMI, Harvard Medical School) led to a number of research contributions to the fields of electron microscopy, membrane biology, water channel physiology, and structural biology. I studied aquaporin-0 (AQP0), the only water channel having two distinct functions: it forms a pore for water permeation, and it creates adhesive junctions in the eye lens. I crystallized AQP0 in two dimensions (2D) for electron crystallography and collected a data set to 1.9-Å resolutionthe highest resolution achieved using this technique, and the highest resolution achieved for any mammalian membrane channel. I developed procedures for molecular replacement and refinementthe first successful implementation of x-ray crystallographic techniques to an electron crystallographic study; this set the stage for using electron crystallography to determine protein structure rapidly.
Our structure, the first atomic resolution structure for an adhesive junction, highlighted how AQP0 membrane channels can mediate the formation of cell-cell membrane junctions. Our study also revealed that the AQP0 water channels close when the protein forms membrane junctions. This allowed us to investigate aspects of water channel regulation at high resolution and furthered our understanding of how these ubiquitous channels function. A striking feature of these 2D crystals is the presence of a continuous lipid bilayer surrounding AQP0. Our membrane model is the first, and only, experimentally determined structure of a eukaryotic membrane surrounding a mammalian protein. The structure allowed us to begin to understand how membrane protein functions may depend on the ability of these proteins to organize the lipid environment around them.
We have now extended our water channel studies to include channel structure in general and channel regulation in particular. AQP0 is regulated by at least three bona fide mechanisms: pH, calmodulin, and phosphorylation.
pH Regulation
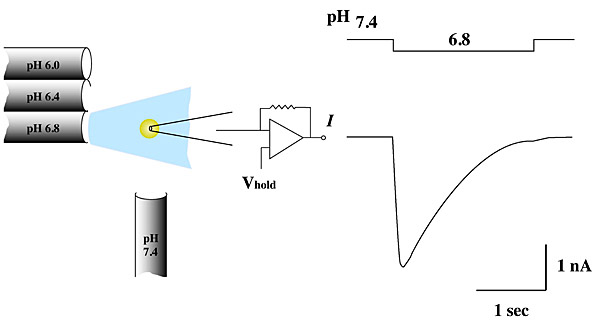
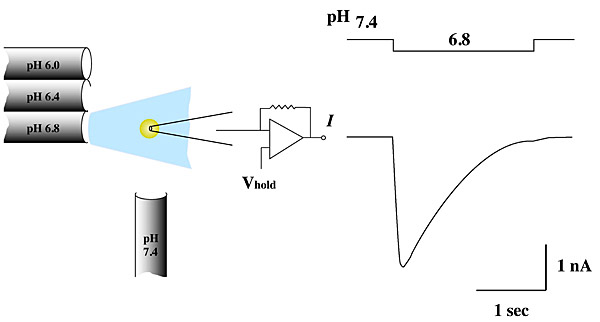
We are studying the structural consequences of AQP0's pH regulation. AQP0's permeability doubles when extracellular pH drops from 7.2 to 6.5 (a physiologically significant event in the eye lens). Two-dimensional crystals are biological membranes in which water channels form functional pores. These crystals are robust enough to allow us to change buffer pH without affecting crystal order, making it possible to use the same 2D crystals to determine AQP0's structure under different conditions. We anticipate that at pH 6.5 the channel will be in a high-permeability state; at pH 7.2, the permeability of AQP0 halves and we expect the channel to be in a low-permeability state.
Regulation by Ca2+/Calmodulin
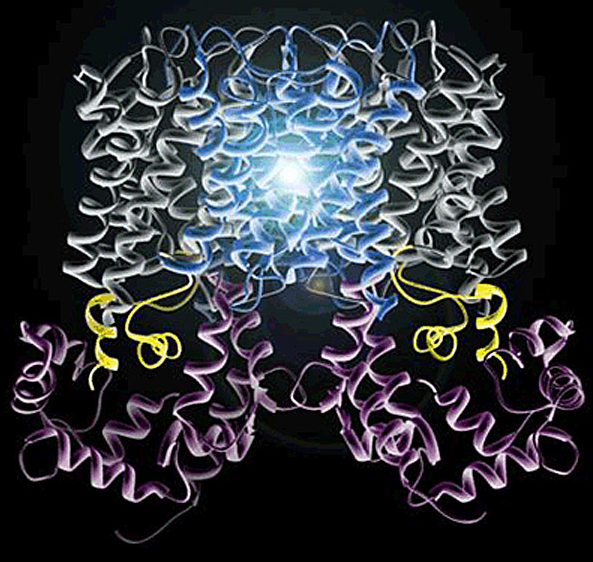
AQP0's permeability is regulated by calmodulin (CaM). Calmodulin is a bilobed Ca2+-binding protein that functions as a ubiquitous secondary messenger in several Ca2+-signaling pathways. CaM modulates the activity of many channels and transporters, including communicating channels, water channels, and voltage-gated cardiac Ca2+ channels. CaM is therefore arguably the most important signaling molecule in the cell, yet no structure of any channel/transporter in complex with CaM has been determined.
We adopted a multidisciplinary approach to structurally characterize the interaction between AQP0 and CaM by NMR, x-ray crystallography, and electron crystallography. Using NMR, we derived a structural model of the AQP0-CaM complex and showed that CaM may act as a channel inhibitor, by capping the water pores of two monomers within the AQP0 tetramer.
Our results suggest that CaM induces cross-cooperativity between monomers within the AQP0 tetramer, and describe for the first time the cooperativity within water channels. For a long time it was unclear why AQPs assemble into tetramers, because each monomer is functionally independent within the tetramer. Our research suggests that the tetramerization is a necessary scaffold for the binding of regulatory proteins such as CaM.
Membrane Transporters: Glucose Transporters
Dietary glucose, the primary source of energy for human cells, is transported from the intestine into the bloodstream and finally into various cell types through glucose transporters (GLUTs), proteins that form specialized pores for the facilitated transport of sugars across biological membranes. The GLUT family belongs to the major facilitator superfamily (MFS) of secondary transportersthe largest group of integral membrane proteins forming pores for transport of diverse substrates such as sugars, drugs, amino acids, and neurotransmitters. Twelve GLUT isoforms have been identified in humans, each having a different expression profile, substrate affinity, and specificity, reflecting the unique glucose requirements of individual tissues and organs.
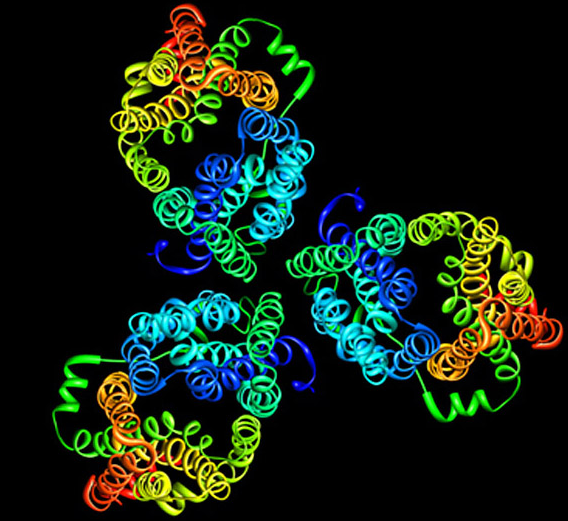
We have expressed, purified, and crystallized the closest bacterial homolog of the human facilitated GLUTthe Escherichia coli H+/galactose symporter (GalP). Surprisingly, the projection structure of GalP indicates that the transporter forms trimers in membranesan oligomerization that is both novel and unexplained. Our functional studies on GalP 2D crystals verify that the membrane-embedded transporter is able to bind substrate. It is possible that trimerization is necessary for protein stability and function, and we are investigating this.
Electrophysiology: Channel Recordings for Structure-Function Analysis
We use electrophysiology and patch-clamping techniques to study the function of channels and transporters. We use the Xenopus oocyte expression system, but we also record channel function from highly ordered 2D crystals for a direct correlation between structure and function of target proteins as they are embedded within a biological membrane.
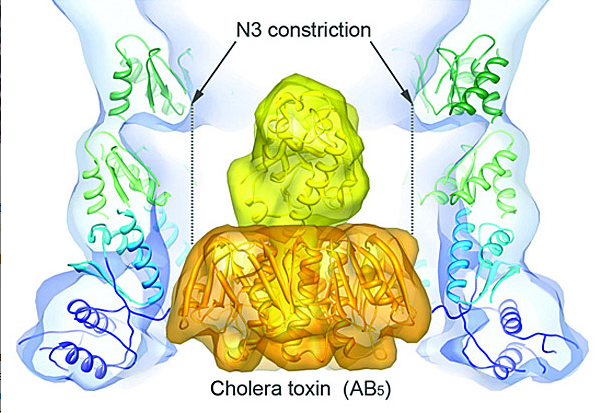
Structural Biology of Cholera Toxin Secretion Channels
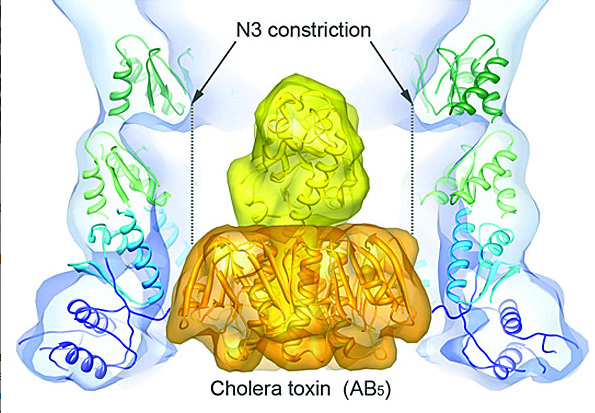
The type II secretion system (T2SS) is a macromolecular complex spanning the inner and outer membranes of Gram-negative bacteria. Remarkably, the T2SS secretes folded proteins, including multimeric assemblies such as cholera toxin from Vibrio cholerae and heat-labile enterotoxin from enterotoxigenic Escherichia coli. The major outer membrane T2SS protein is the secretin GspD. Using cryo-EM and single-particle reconstruction techniques, we are investigating the structure and function of this cholera toxin secretion channel. Cryo-EM reconstruction of the V. cholerae secretin at 19-Å resolution revealed a dodecameric structure reminiscent of a barrel, with a large channel at its center that contains a closed periplasmic gate. The GspD periplasmic domain forms a vestibule with a conserved constriction, and it binds to a pentameric exoprotein and to the trimeric tip of the T2SS pseudopilus. By combining our results with structures of the cholera toxin and T2SS pseudopilus, we provide a structural basis for a possible secretion mechanism of the T2SS. We are working to improve the resolution of our maps.
Method Development: High-Throughput Electron Crystallography
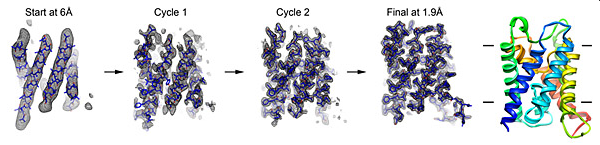
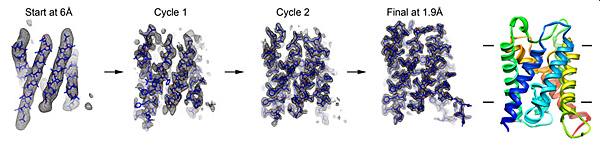
In electron crystallography, membrane protein structure is determined from 2D crystals where the protein is embedded in a membrane. Once large and well-ordered 2D crystals are grown, one of the bottlenecks in electron crystallography is the collection of image data to provide experimental phases directly to high resolution. We recently developed a new approach to bypass this bottleneck, eliminating the need for high-resolution imaging. We used the strengths of electron crystallography to rapidly obtain accurate experimental-phase information from low-resolution images and to obtain accurate high-resolution amplitude information from electron diffraction. The low-resolution experimental phases were used for the placement of ?-helix fragments and were extended to high resolution using phases from the fragments. Phases were further improved by density modifications, followed by fragment expansion and structure refinement against the high-resolution diffraction data. Using this approach, we determined structures of three membrane proteins rapidly and accurately to atomic resolution without high-resolution image data. We are working to fully automate this method.
As of October 07, 2011