Filter
Associated Lab
- Ahrens Lab (4) Apply Ahrens Lab filter
- Betzig Lab (1) Apply Betzig Lab filter
- Beyene Lab (1) Apply Beyene Lab filter
- Druckmann Lab (1) Apply Druckmann Lab filter
- Harris Lab (4) Apply Harris Lab filter
- Hermundstad Lab (1) Apply Hermundstad Lab filter
- Jayaraman Lab (9) Apply Jayaraman Lab filter
- Karpova Lab (1) Apply Karpova Lab filter
- Keller Lab (1) Apply Keller Lab filter
- Lavis Lab (8) Apply Lavis Lab filter
- Leonardo Lab (1) Apply Leonardo Lab filter
- Liu (Zhe) Lab (1) Apply Liu (Zhe) Lab filter
- Looger Lab (24) Apply Looger Lab filter
- Podgorski Lab (5) Apply Podgorski Lab filter
- Rubin Lab (1) Apply Rubin Lab filter
- Schreiter Lab (67) Apply Schreiter Lab filter
- Stringer Lab (1) Apply Stringer Lab filter
- Svoboda Lab (13) Apply Svoboda Lab filter
- Tillberg Lab (1) Apply Tillberg Lab filter
- Turner Lab (4) Apply Turner Lab filter
- Zlatic Lab (1) Apply Zlatic Lab filter
Associated Project Team
Publication Date
- 2025 (3) Apply 2025 filter
- 2024 (5) Apply 2024 filter
- 2023 (6) Apply 2023 filter
- 2021 (1) Apply 2021 filter
- 2020 (5) Apply 2020 filter
- 2019 (4) Apply 2019 filter
- 2018 (4) Apply 2018 filter
- 2017 (4) Apply 2017 filter
- 2016 (2) Apply 2016 filter
- 2015 (4) Apply 2015 filter
- 2013 (7) Apply 2013 filter
- 2012 (2) Apply 2012 filter
- 2011 (3) Apply 2011 filter
- 2010 (2) Apply 2010 filter
- 2009 (3) Apply 2009 filter
- 2008 (3) Apply 2008 filter
- 2007 (3) Apply 2007 filter
- 2006 (2) Apply 2006 filter
- 2003 (1) Apply 2003 filter
- 2001 (2) Apply 2001 filter
- 1999 (1) Apply 1999 filter
Type of Publication
67 Publications
Showing 51-60 of 67 resultsEscherichia coli NikR regulates cellular nickel uptake by binding to the nik operon in the presence of nickel and blocking transcription of genes encoding the nickel uptake transporter. NikR has two binding affinities for the nik operon: a nanomolar dissociation constant with stoichiometric nickel and a picomolar dissociation constant with excess nickel [Bloom, S. L., and Zamble, D. B. (2004) Biochemistry 43, 10029-10038; Chivers, P. T., and Sauer, R. T. (2002) Chem. Biol. 9, 1141-1148]. While it is known that the stoichiometric nickel ions bind at the NikR tetrameric interface [Schreiter, E. R., et al. (2003) Nat. Struct. Biol. 10, 794-799; Schreiter, E. R., et al. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 13676-13681], the binding sites for excess nickel ions have not been fully described. Here we have determined the crystal structure of NikR in the presence of excess nickel to 2.6 A resolution and have obtained nickel anomalous data (1.4845 A) in the presence of excess nickel for both NikR alone and NikR cocrystallized with a 30-nucleotide piece of double-stranded DNA containing the nik operon. These anomalous data show that excess nickel ions do not bind to a single location on NikR but instead reveal a total of 22 possible low-affinity nickel sites on the NikR tetramer. These sites, for which there are six different types, are all on the surface of NikR, and most are found in both the NikR alone and NikR-DNA structures. Using a combination of crystallographic data and molecular dynamics simulations, the nickel sites can be described as preferring octahedral geometry, utilizing one to three protein ligands (typically histidine) and at least two water molecules.
Heparin-binding EGF-like growth factor (HB-EGF) is a ligand for EGF receptor (EGFR) and possesses the ability to signal in juxtacrine, autocrine and/or paracrine mode, with these alternatives being governed by the degree of proteolytic release of the ligand. Although the spatial range of diffusion of released HB-EGF is restricted by binding heparan-sulfate proteoglycans (HSPGs) in the extracellular matrix and/or cellular glycocalyx, ascertaining mechanisms governing non-released HB-EGF localization is also important for understanding its effects. We have employed a new method for independently tracking the localization of the extracellular EGF-like domain of HB-EGF and the cytoplasmic C-terminus. A striking observation was the absence of the HB-EGF transmembrane pro-form from the leading edge of COS-7 cells in a wound-closure assay; instead, this protein localized in regions of cell-cell contact. A battery of detailed experiments found that this localization derives from a trans interaction between extracellular HSPGs and the HB-EGF heparin-binding domain, and that disruption of this interaction leads to increased release of soluble ligand and a switch in cell phenotype from juxtacrine-induced growth inhibition to autocrine-induced proliferation. Our results indicate that extracellular HSPGs serve to sequester the transmembrane pro-form of HB-EGF at the point of cell-cell contact, and that this plays a role in governing the balance between juxtacrine versus autocrine and paracrine signaling.
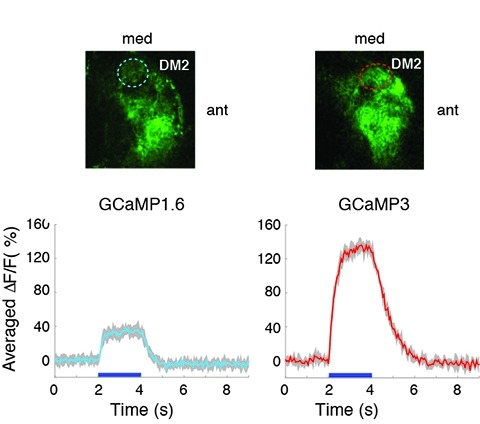
Genetically encoded calcium indicators (GECIs) can be used to image activity in defined neuronal populations. However, current GECIs produce inferior signals compared to synthetic indicators and recording electrodes, precluding detection of low firing rates. We developed a single-wavelength GCaMP2-based GECI (GCaMP3), with increased baseline fluorescence (3-fold), increased dynamic range (3-fold) and higher affinity for calcium (1.3-fold). We detected GCaMP3 fluorescence changes triggered by single action potentials in pyramidal cell dendrites, with signal-to-noise ratio and photostability substantially better than those of GCaMP2, D3cpVenus and TN-XXL. In Caenorhabditis elegans chemosensory neurons and the Drosophila melanogaster antennal lobe, sensory stimulation-evoked fluorescence responses were significantly enhanced with GCaMP3 (4-6-fold). In somatosensory and motor cortical neurons in the intact mouse, GCaMP3 detected calcium transients with amplitudes linearly dependent on action potential number. Long-term imaging in the motor cortex of behaving mice revealed large fluorescence changes in imaged neurons over months.
Thioredoxin-interacting protein (Txnip), originally characterized as an inhibitor of thioredoxin, is now known to be a critical regulator of glucose metabolism in vivo. Txnip is a member of the alpha-arrestin protein family; the alpha-arrestins are related to the classical beta-arrestins and visual arrestins. Txnip is the only alpha-arrestin known to bind thioredoxin, and it is not known whether the metabolic effects of Txnip are related to its ability to bind thioredoxin or related to conserved alpha-arrestin function. Here we show that wild type Txnip and Txnip C247S, a Txnip mutant that does not bind thioredoxin in vitro, both inhibit glucose uptake in mature adipocytes and in primary skin fibroblasts. Furthermore, we show that Txnip C247S does not bind thioredoxin in cells, using thiol alkylation to trap the Txnip-thioredoxin complex. Because Txnip function was independent of thioredoxin binding, we tested whether inhibition of glucose uptake was conserved in the related alpha-arrestins Arrdc4 and Arrdc3. Both Txnip and Arrdc4 inhibited glucose uptake and lactate output, while Arrdc3 had no effect. Structure-function analysis indicated that Txnip and Arrdc4 inhibit glucose uptake independent of the C-terminal WW-domain binding motifs, recently identified as important in yeast alpha-arrestins. Instead, regulation of glucose uptake was intrinsic to the arrestin domains themselves. These data demonstrate that Txnip regulates cellular metabolism independent of its binding to thioredoxin and reveal the arrestin domains as crucial structural elements in metabolic functions of alpha-arrestin proteins.
The genetically encoded calcium indicator GCaMP2 shows promise for neural network activity imaging, but is currently limited by low signal-to-noise ratio. We describe x-ray crystal structures as well as solution biophysical and spectroscopic characterization of GCaMP2 in the calcium-free dark state, and in two calcium-bound bright states: a monomeric form that dominates at intracellular concentrations observed during imaging experiments and an unexpected domain-swapped dimer with decreased fluorescence. This series of structures provides insight into the mechanism of Ca2+-induced fluorescence change. Upon calcium binding, the calmodulin (CaM) domain wraps around the M13 peptide, creating a new domain interface between CaM and the circularly permuted enhanced green fluorescent protein domain. Residues from CaM alter the chemical environment of the circularly permuted enhanced green fluorescent protein chromophore and, together with flexible inter-domain linkers, block solvent access to the chromophore. Guided by the crystal structures, we engineered a series of GCaMP2 point mutants to probe the mechanism of GCaMP2 function and characterized one mutant with significantly improved signal-to-noise. The mutation is located at a domain interface and its effect on sensor function could not have been predicted in the absence of structural data.
Fluorescent proteins and their engineered variants have played an important role in the study of biology. The genetically encoded calcium-indicator protein GCaMP2 comprises a circularly permuted fluorescent protein coupled to the calcium-binding protein calmodulin and a calmodulin target peptide, M13, derived from the intracellular calmodulin target myosin light-chain kinase and has been used to image calcium transients in vivo. To aid rational efforts to engineer improved variants of GCaMP2, this protein was crystallized in the calcium-saturated form. X-ray diffraction data were collected to 2.0 A resolution. The crystals belong to space group C2, with unit-cell parameters a = 126.1.
Chlamydomonas reinhardtii centrin is a member of the EF-hand calcium-binding superfamily. It is found in the basal body complex and is important for flagellar motility. Like other members of the EF-hand family, centrin interacts with and modulates the function of other proteins in a calcium-dependent manner. To understand how C. reinhardtii centrin interacts with its protein targets, it has been crystallized in the presence of the model peptide melittin and X-ray diffraction data have been collected to 2.2 A resolution. The crystals are orthorhombic, with unit-cell parameters a = 52.1, b = 114.4, c = 34.8 A, and are likely to belong to space group P2(1)2(1)2.
In the presence of excess nickel, Escherichia coli NikR regulates cellular nickel uptake by suppressing the transcription of the nik operon, which encodes the nickel uptake transporter, NikABCDE. Previously published in vitro studies have shown that NikR is capable of binding a range of divalent transition metal ions in addition to Ni2+, including Co2+, Cu2+, Zn2+, and Cd2+. To understand how the high-affinity nickel binding site of NikR is able to accommodate these other metal ions, and to improve our understanding of NikR's mechanism of binding to DNA, we have determined structures of the metal-binding domain (MBD) of NikR in the apo form and in complex with Cu2+ and Zn2+ ions and compared them with the previously published structures with Ni2+. We observe that Cu2+ ions bind in a manner very similar to that of Ni2+, with a square planar geometry but with longer bond lengths. Crystals grown in the presence of Zn2+ reveal a protein structure similar to that of apo MBD with a disordered alpha3 helix, but with two electron density peaks near the Ni2+ binding site corresponding to two Zn2+ ions. These structural findings along with biochemical data on NikR support a hypothesis that ordering of the alpha3 helix is important for repressor activation.
The ribbon-helix-helix (RHH) superfamily of transcription factors uses a conserved three-dimensional structural motif to bind to DNA in a sequence-specific manner. This functionally diverse protein superfamily regulates the transcription of genes that are involved in the uptake of metals, amino-acid biosynthesis, cell division, the control of plasmid copy number, the lytic cycle of bacteriophages and, perhaps, many other cellular processes. In this Analysis, the structures of different RHH transcription factors are compared in order to evaluate the sequence motifs that are required for RHH-domain folding and DNA binding, as well as to identify conserved protein-DNA interactions in this superfamily.
S-nitrosylation is a post-translational protein modification that can alter the function of a variety of proteins. Despite the growing wealth of information that this modification may have important functional consequences, little is known about the structure of the moiety or its effect on protein tertiary structure. Here we report high-resolution x-ray crystal structures of S-nitrosylated and unmodified blackfin tuna myoglobin, which demonstrate that in vitro S-nitrosylation of this protein at the surface-exposed Cys-10 directly causes a reversible conformational change by "wedging" apart a helix and loop. Furthermore, we have demonstrated in solution and in a single crystal that reduction of the S-nitrosylated myoglobin with dithionite results in NO cleavage from the sulfur of Cys-10 and rebinding to the reduced heme iron, showing the reversibility of both the modification and the conformational changes. Finally, we report the 0.95-A structure of ferrous nitrosyl myoglobin, which provides an accurate structural view of the NO coordination geometry in the context of a globin heme pocket.