Filter
Associated Lab
- Ahrens Lab (7) Apply Ahrens Lab filter
- Druckmann Lab (2) Apply Druckmann Lab filter
- Dudman Lab (1) Apply Dudman Lab filter
- Freeman Lab (2) Apply Freeman Lab filter
- Harris Lab (7) Apply Harris Lab filter
- Hermundstad Lab (1) Apply Hermundstad Lab filter
- Hess Lab (2) Apply Hess Lab filter
- Jayaraman Lab (10) Apply Jayaraman Lab filter
- Karpova Lab (2) Apply Karpova Lab filter
- Keller Lab (5) Apply Keller Lab filter
- Lavis Lab (8) Apply Lavis Lab filter
- Leonardo Lab (4) Apply Leonardo Lab filter
- Remove Looger Lab filter Looger Lab
- Podgorski Lab (6) Apply Podgorski Lab filter
- Rubin Lab (2) Apply Rubin Lab filter
- Schreiter Lab (24) Apply Schreiter Lab filter
- Simpson Lab (1) Apply Simpson Lab filter
- Spruston Lab (1) Apply Spruston Lab filter
- Sternson Lab (2) Apply Sternson Lab filter
- Svoboda Lab (20) Apply Svoboda Lab filter
- Tervo Lab (1) Apply Tervo Lab filter
- Tillberg Lab (1) Apply Tillberg Lab filter
- Turner Lab (1) Apply Turner Lab filter
- Zlatic Lab (1) Apply Zlatic Lab filter
Associated Project Team
Publication Date
- 2024 (1) Apply 2024 filter
- 2023 (5) Apply 2023 filter
- 2022 (7) Apply 2022 filter
- 2021 (11) Apply 2021 filter
- 2020 (7) Apply 2020 filter
- 2019 (15) Apply 2019 filter
- 2018 (8) Apply 2018 filter
- 2017 (6) Apply 2017 filter
- 2016 (10) Apply 2016 filter
- 2015 (9) Apply 2015 filter
- 2014 (11) Apply 2014 filter
- 2013 (10) Apply 2013 filter
- 2012 (13) Apply 2012 filter
- 2011 (7) Apply 2011 filter
- 2010 (7) Apply 2010 filter
- 2009 (7) Apply 2009 filter
- 2008 (3) Apply 2008 filter
Type of Publication
137 Publications
Showing 21-30 of 137 resultsHuman leukocyte antigen (HLA) is a key genetic factor conferring risk of systemic lupus erythematosus (SLE), but precise independent localization of HLA effects is extremely challenging. As a result, the contribution of specific HLA alleles and amino-acid residues to the overall risk of SLE and to risk of specific autoantibodies are far from completely understood. Here, we dissected (a) overall SLE association signals across HLA, (b) HLA-peptide interaction, and (c) residue-autoantibody association. Classical alleles, SNPs, and amino-acid residues of eight HLA genes were imputed across 4,915 SLE cases and 13,513 controls from Eastern Asia. We performed association followed by conditional analysis across HLA, assessing both overall SLE risk and risk of autoantibody production. DR15 alleles HLA-DRB1*15:01 (P = 1.4x10-27, odds ratio (OR) = 1.57) and HLA-DQB1*06:02 (P = 7.4x10-23, OR = 1.55) formed the most significant haplotype (OR = 2.33). Conditioned protein-residue signals were stronger than allele signals and mapped predominantly to HLA-DRB1 residue 13 (P = 2.2x10-75) and its proxy position 11 (P = 1.1x10-67), followed by HLA-DRB1-37 (P = 4.5x10-24). After conditioning on HLA-DRB1, novel associations at HLA-A-70 (P = 1.4x10-8), HLA-DPB1-35 (P = 9.0x10-16), HLA-DQB1-37 (P = 2.7x10-14), and HLA-B-9 (P = 6.5x10-15) emerged. Together, these seven residues increased the proportion of explained heritability due to HLA to 2.6%. Risk residues for both overall disease and hallmark autoantibodies (i.e., nRNP: DRB1-11, P = 2.0x10-14; DRB1-13, P = 2.9x10-13; DRB1-30, P = 3.9x10-14) localized to the peptide-binding groove of HLA-DRB1. Enrichment for specific amino-acid characteristics in the peptide-binding groove correlated with overall SLE risk and with autoantibody presence. Risk residues were in primarily negatively charged side-chains, in contrast with rheumatoid arthritis. We identified novel SLE signals in HLA Class I loci (HLA-A, HLA-B), and localized primary Class II signals to five residues in HLA-DRB1, HLA-DPB1, and HLA-DQB1. These findings provide insights about the mechanisms by which the risk residues interact with each other to produce autoantibodies and are involved in SLE pathophysiology.
We describe an intensity-based glutamate-sensing fluorescent reporter (iGluSnFR) with signal-to-noise ratio and kinetics appropriate for in vivo imaging. We engineered iGluSnFR in vitro to maximize its fluorescence change, and we validated its utility for visualizing glutamate release by neurons and astrocytes in increasingly intact neurological systems. In hippocampal culture, iGluSnFR detected single field stimulus-evoked glutamate release events. In pyramidal neurons in acute brain slices, glutamate uncaging at single spines showed that iGluSnFR responds robustly and specifically to glutamate in situ, and responses correlate with voltage changes. In mouse retina, iGluSnFR-expressing neurons showed intact light-evoked excitatory currents, and the sensor revealed tonic glutamate signaling in response to light stimuli. In worms, glutamate signals preceded and predicted postsynaptic calcium transients. In zebrafish, iGluSnFR revealed spatial organization of direction-selective synaptic activity in the optic tectum. Finally, in mouse forelimb motor cortex, iGluSnFR expression in layer V pyramidal neurons revealed task-dependent single-spine activity during running.
The target for the "rapid" (<24 h) antidepressant effects of S-ketamine is unknown, vitiating programs to rationally develop more effective rapid antidepressants. To describe a drug's target, one must first understand the compartments entered by the drug, at all levels-the organ, the cell, and the organelle. We have, therefore, developed molecular tools to measure the subcellular, organellar pharmacokinetics of S-ketamine. The tools are genetically encoded intensity-based S-ketamine-sensing fluorescent reporters, iSKetSnFR1 and iSKetSnFR2. In solution, these biosensors respond to S-ketamine with a sensitivity, S-slope = delta(F/F)/(delta[S-ketamine]) of 0.23 and 1.9/μM, respectively. The iSKetSnFR2 construct allows measurements at <0.3 μM S-ketamine. The iSKetSnFR1 and iSKetSnFR2 biosensors display >100-fold selectivity over other ligands tested, including R-ketamine. We targeted each of the sensors to either the plasma membrane (PM) or the endoplasmic reticulum (ER). Measurements on these biosensors expressed in Neuro2a cells and in human dopaminergic neurons differentiated from induced pluripotent stem cells (iPSCs) show that S-ketamine enters the ER within a few seconds after appearing in the external solution near the PM, then leaves as rapidly after S-ketamine is removed from the extracellular solution. In cells, S-slopes for the ER and PM-targeted sensors differ by <2-fold, indicating that the ER [S-ketamine] is less than 2-fold different from the extracellular [S-ketamine]. Organelles represent potential compartments for the engagement of S-ketamine with its antidepressant target, and potential S-ketamine targets include organellar ion channels, receptors, and transporters.
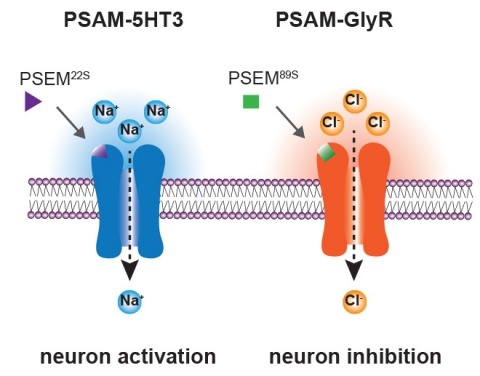
Ionic flux mediates essential physiological and behavioral functions in defined cell populations. Cell type-specific activators of diverse ionic conductances are needed for probing these effects. We combined chemistry and protein engineering to enable the systematic creation of a toolbox of ligand-gated ion channels (LGICs) with orthogonal pharmacologic selectivity and divergent functional properties. The LGICs and their small-molecule effectors were able to activate a range of ionic conductances in genetically specified cell types. LGICs constructed for neuronal perturbation could be used to selectively manipulate neuron activity in mammalian brains in vivo. The diversity of ion channel tools accessible from this approach will be useful for examining the relationship between neuronal activity and animal behavior, as well as for cell biological and physiological applications requiring chemical control of ion conductance.
We report the rational engineering of a remarkably stable yellow fluorescent protein (YFP), 'hyperfolder YFP' (hfYFP), that withstands chaotropic conditions that denature most biological structures within seconds, including superfolder green fluorescent protein (GFP). hfYFP contains no cysteines, is chloride insensitive and tolerates aldehyde and osmium tetroxide fixation better than common fluorescent proteins, enabling its use in expansion and electron microscopies. We solved crystal structures of hfYFP (to 1.7-Å resolution), a monomeric variant, monomeric hyperfolder YFP (1.6 Å) and an mGreenLantern mutant (1.2 Å), and then rationally engineered highly stable 405-nm-excitable GFPs, large Stokes shift (LSS) monomeric GFP (LSSmGFP) and LSSA12 from these structures. Lastly, we directly exploited the chemical stability of hfYFP and LSSmGFP by devising a fluorescence-assisted protein purification strategy enabling all steps of denaturing affinity chromatography to be visualized using ultraviolet or blue light. hfYFP and LSSmGFP represent a new generation of robustly stable fluorescent proteins developed for advanced biotechnological applications.
The R‐specific alcohol dehydrogenase from Lactobacillus brevis (Lb‐ADH) catalyzes the enantioselective reduction of prochiral ketones to the corresponding secondary alcohols. It is stable and has broad substrate specificity. These features make this enzyme an attractive candidate for biotechnological applications. A drawback is its preference for NADP(H) as a cofactor, which is more expensive and labile than NAD(H). Structure‐based computational protein engineering was used to predict mutations to alter the cofactor specificity of Lb‐ADH. Mutations were introduced into Lb‐ADH and tested against the substrate acetophenone, with either NAD(H) or NADP(H) as cofactor. The mutant Arg38Pro showed fourfold increased activity with acetophenone and NAD(H) relative to the wild type. Both Arg38Pro and wild type exhibit a pH optimum of 5.5 with NAD(H) as cofactor, significantly more acidic than with NADP(H). These and related Lb‐ADH mutants may prove useful for the green synthesis of pharmaceutical precursors.
Integrin alpha M (ITGAM; CD11b) is a component of the macrophage-1 antigen complex, which mediates leukocyte adhesion, migration and phagocytosis as part of the immune system. We previously identified a missense polymorphism, rs1143679 (R77H), strongly associated with systemic lupus erythematosus (SLE). However, the molecular mechanisms of this variant are incompletely understood. A meta-analysis of published and novel data on 28 439 individuals with European, African, Hispanic and Asian ancestries reinforces genetic association between rs1143679 and SLE [Pmeta = 3.60 × 10(-90), odds ratio (OR) = 1.76]. Since rs1143679 is in the most active region of chromatin regulation and transcription factor binding in ITGAM, we quantitated ITGAM RNA and surface protein levels in monocytes from patients with each rs1143679 genotype. We observed that transcript levels significantly decreased for the risk allele ('A') relative to the non-risk allele ('G'), in a dose-dependent fashion: ('AA' < 'AG' < 'GG'). CD11b protein levels in patients' monocytes were directly correlated with RNA levels. Strikingly, heterozygous individuals express much lower (average 10- to 15-fold reduction) amounts of the 'A' transcript than 'G' transcript. We found that the non-risk sequence surrounding rs1143679 exhibits transcriptional enhancer activity in vivo and binds to Ku70/80, NFKB1 and EBF1 in vitro, functions that are significantly reduced with the risk allele. Mutant CD11b protein shows significantly reduced binding to fibrinogen and vitronectin, relative to non-risk, both in purified protein and in cellular models. This two-pronged contribution (nucleic acid- and protein-level) of the rs1143679 risk allele to decreasing ITGAM activity provides insight into the molecular mechanisms of its potent association with SLE.
The spatiotemporal activities of astrocyte Ca(2+) signaling in mature neuronal circuits remain unclear. We used genetically encoded Ca(2+) and glutamate indicators as well as pharmacogenetic and electrical control of neurotransmitter release to explore astrocyte activity in the hippocampal mossy fiber pathway. Our data revealed numerous localized, spontaneous Ca(2+) signals in astrocyte branches and territories, but these were not driven by neuronal activity or glutamate. Moreover, evoked astrocyte Ca(2+) signaling changed linearly with the number of mossy fiber action potentials. Under these settings, astrocyte responses were global, suppressed by neurotransmitter clearance, and mediated by glutamate and GABA. Thus, astrocyte engagement in the fully developed mossy fiber pathway was slow and territorial, contrary to that frequently proposed for astrocytes within microcircuits. We show that astrocyte Ca(2+) signaling functionally segregates large volumes of neuropil and that these transients are not suited for responding to, or regulating, single synapses in the mossy fiber pathway.
We recently identified ten novel SLE susceptibility loci in Asians and uncovered several additional suggestive loci requiring further validation. This study aimed to replicate five of these suggestive loci in a Han Chinese cohort from Hong Kong, followed by meta-analysis (11,656 cases and 23,968 controls) on previously reported Asian and European populations, and to perform bioinformatic analyses on all 82 reported SLE loci to identify shared regulatory signatures. We performed a battery of analyses for these five loci, as well as joint analyses on all 82 SLE loci. All five loci passed genome-wide significance: MYNN (rs10936599, Pmeta = 1.92 × 10-13, OR = 1.14), ATG16L2 (rs11235604, Pmeta = 8.87 × 10 -12, OR = 0.78), CCL22 (rs223881, Pmeta = 5.87 × 10-16, OR = 0.87), ANKS1A (rs2762340, Pmeta = 4.93 × 10-15, OR = 0.87) and RNASEH2C (rs1308020, Pmeta = 2.96 × 10-19, OR = 0.84) and co-located with annotated gene regulatory elements. The novel loci share genetic signatures with other reported SLE loci, including effects on gene expression, transcription factor binding, and epigenetic characteristics. Most (56%) of the correlated (r2 > 0.8) SNPs from the 82 SLE loci were implicated in differential expression (9.81 × 10-198 < P < 5 × 10-3) of cis-genes. Transcription factor binding sites for p53, MEF2A and E2F1 were significantly (P < 0.05) over-represented in SLE loci, consistent with apoptosis playing a critical role in SLE. Enrichment analysis revealed common pathways, gene ontology, protein domains, and cell type-specific expression. In summary, we provide evidence of five novel SLE susceptibility loci. Integrated bioinformatics using all 82 loci revealed that SLE susceptibility loci share many gene regulatory features, suggestive of conserved mechanisms of SLE etiopathogenesis.
The genetically encoded calcium indicator GCaMP2 shows promise for neural network activity imaging, but is currently limited by low signal-to-noise ratio. We describe x-ray crystal structures as well as solution biophysical and spectroscopic characterization of GCaMP2 in the calcium-free dark state, and in two calcium-bound bright states: a monomeric form that dominates at intracellular concentrations observed during imaging experiments and an unexpected domain-swapped dimer with decreased fluorescence. This series of structures provides insight into the mechanism of Ca2+-induced fluorescence change. Upon calcium binding, the calmodulin (CaM) domain wraps around the M13 peptide, creating a new domain interface between CaM and the circularly permuted enhanced green fluorescent protein domain. Residues from CaM alter the chemical environment of the circularly permuted enhanced green fluorescent protein chromophore and, together with flexible inter-domain linkers, block solvent access to the chromophore. Guided by the crystal structures, we engineered a series of GCaMP2 point mutants to probe the mechanism of GCaMP2 function and characterized one mutant with significantly improved signal-to-noise. The mutation is located at a domain interface and its effect on sensor function could not have been predicted in the absence of structural data.