Filter
Associated Lab
- Ahrens Lab (5) Apply Ahrens Lab filter
- Aso Lab (1) Apply Aso Lab filter
- Baker Lab (2) Apply Baker Lab filter
- Betzig Lab (4) Apply Betzig Lab filter
- Bock Lab (2) Apply Bock Lab filter
- Cardona Lab (1) Apply Cardona Lab filter
- Cui Lab (2) Apply Cui Lab filter
- Dickson Lab (3) Apply Dickson Lab filter
- Druckmann Lab (1) Apply Druckmann Lab filter
- Dudman Lab (2) Apply Dudman Lab filter
- Eddy/Rivas Lab (2) Apply Eddy/Rivas Lab filter
- Egnor Lab (1) Apply Egnor Lab filter
- Fetter Lab (3) Apply Fetter Lab filter
- Fitzgerald Lab (1) Apply Fitzgerald Lab filter
- Gonen Lab (11) Apply Gonen Lab filter
- Grigorieff Lab (4) Apply Grigorieff Lab filter
- Harris Lab (3) Apply Harris Lab filter
- Heberlein Lab (6) Apply Heberlein Lab filter
- Hermundstad Lab (1) Apply Hermundstad Lab filter
- Hess Lab (2) Apply Hess Lab filter
- Jayaraman Lab (3) Apply Jayaraman Lab filter
- Ji Lab (1) Apply Ji Lab filter
- Johnson Lab (1) Apply Johnson Lab filter
- Karpova Lab (1) Apply Karpova Lab filter
- Keller Lab (10) Apply Keller Lab filter
- Lavis Lab (4) Apply Lavis Lab filter
- Leonardo Lab (3) Apply Leonardo Lab filter
- Lippincott-Schwartz Lab (11) Apply Lippincott-Schwartz Lab filter
- Looger Lab (10) Apply Looger Lab filter
- Magee Lab (3) Apply Magee Lab filter
- Menon Lab (3) Apply Menon Lab filter
- Pachitariu Lab (3) Apply Pachitariu Lab filter
- Pavlopoulos Lab (1) Apply Pavlopoulos Lab filter
- Reiser Lab (2) Apply Reiser Lab filter
- Riddiford Lab (5) Apply Riddiford Lab filter
- Romani Lab (1) Apply Romani Lab filter
- Rubin Lab (5) Apply Rubin Lab filter
- Satou Lab (2) Apply Satou Lab filter
- Scheffer Lab (3) Apply Scheffer Lab filter
- Schreiter Lab (7) Apply Schreiter Lab filter
- Sgro Lab (1) Apply Sgro Lab filter
- Singer Lab (9) Apply Singer Lab filter
- Spruston Lab (2) Apply Spruston Lab filter
- Stern Lab (6) Apply Stern Lab filter
- Sternson Lab (3) Apply Sternson Lab filter
- Svoboda Lab (10) Apply Svoboda Lab filter
- Tjian Lab (1) Apply Tjian Lab filter
- Truman Lab (3) Apply Truman Lab filter
- Turaga Lab (2) Apply Turaga Lab filter
- Turner Lab (2) Apply Turner Lab filter
- Wu Lab (3) Apply Wu Lab filter
- Zlatic Lab (2) Apply Zlatic Lab filter
Associated Project Team
Publication Date
- December 2013 (13) Apply December 2013 filter
- November 2013 (10) Apply November 2013 filter
- October 2013 (20) Apply October 2013 filter
- September 2013 (19) Apply September 2013 filter
- August 2013 (15) Apply August 2013 filter
- July 2013 (19) Apply July 2013 filter
- June 2013 (17) Apply June 2013 filter
- May 2013 (10) Apply May 2013 filter
- April 2013 (12) Apply April 2013 filter
- March 2013 (11) Apply March 2013 filter
- February 2013 (19) Apply February 2013 filter
- January 2013 (29) Apply January 2013 filter
- Remove 2013 filter 2013
Type of Publication
194 Publications
Showing 31-40 of 194 resultsBACKGROUND: The insect brain can be divided into neuropils that are formed by neurites of both local and remote origin. The complexity of the interconnections obscures how these neuropils are established and interconnected through development. The Drosophila central brain develops from a fixed number of neuroblasts (NBs) that deposit neurons in regional clusters. RESULTS: By determining individual NB clones and pursuing their projections into specific neuropils, we unravel the regional development of the brain neural network. Exhaustive clonal analysis revealed 95 stereotyped neuronal lineages with characteristic cell-body locations and neurite trajectories. Most clones show complex projection patterns, but despite the complexity, neighboring clones often coinnervate the same local neuropil or neuropils and further target a restricted set of distant neuropils. CONCLUSIONS: These observations argue for regional clonal development of both neuropils and neuropil connectivity throughout the Drosophila central brain.
View Publication PageThe Influenza A virus genome consists of eight negative sense, single-stranded RNA segments. Although it has been established that most virus particles contain a single copy of each of the eight viral RNAs, the packaging selection mechanism remains poorly understood. Influenza viral RNAs are synthesized in the nucleus, exported into the cytoplasm and travel to the plasma membrane where viral budding and genome packaging occurs. Due to the difficulties in analyzing associated vRNPs while preserving information about their positions within the cell, it has remained unclear how and where during cellular trafficking the viral RNAs of different segments encounter each other. Using a multicolor single-molecule sensitivity fluorescence in situ hybridization (smFISH) approach, we have quantitatively monitored the colocalization of pairs of influenza viral RNAs in infected cells. We found that upon infection, the viral RNAs from the incoming particles travel together until they reach the nucleus. The viral RNAs were then detected in distinct locations in the nucleus; they are then exported individually and initially remain separated in the cytoplasm. At later time points, the different viral RNA segments gather together in the cytoplasm in a microtubule independent manner. Viral RNAs of different identities colocalize at a high frequency when they are associated with Rab11 positive vesicles, suggesting that Rab11 positive organelles may facilitate the association of different viral RNAs. Using engineered influenza viruses lacking the expression of HA or M2 protein, we showed that these viral proteins are not essential for the colocalization of two different viral RNAs in the cytoplasm. In sum, our smFISH results reveal that the viral RNAs travel together in the cytoplasm before their arrival at the plasma membrane budding sites. This newly characterized step of the genome packaging process demonstrates the precise spatiotemporal regulation of the infection cycle.
The suppression of tumorigenicity 2/IL-33 (ST2/IL-33) pathway has been implicated in several immune and inflammatory diseases. ST2 is produced as 2 isoforms. The membrane-bound isoform (ST2L) induces an immune response when bound to its ligand, IL-33. The other isoform is a soluble protein (sST2) that is thought to be a decoy receptor for IL-33 signaling. Elevated sST2 levels in serum are associated with an increased risk for cardiovascular disease. We investigated the determinants of sST2 plasma concentrations in 2,991 Framingham Offspring Cohort participants. While clinical and environmental factors explained some variation in sST2 levels, much of the variation in sST2 production was driven by genetic factors. In a genome-wide association study (GWAS), multiple SNPs within IL1RL1 (the gene encoding ST2) demonstrated associations with sST2 concentrations. Five missense variants of IL1RL1 correlated with higher sST2 levels in the GWAS and mapped to the intracellular domain of ST2, which is absent in sST2. In a cell culture model, IL1RL1 missense variants increased sST2 expression by inducing IL-33 expression and enhancing IL-33 responsiveness (via ST2L). Our data suggest that genetic variation in IL1RL1 can result in increased levels of sST2 and alter immune and inflammatory signaling through the ST2/IL-33 pathway.
The neural circuits that mediate behavioral choice evaluate and integrate information from the environment with internal demands and then initiate a behavioral response. Even circuits that support simple decisions remain poorly understood. In Drosophila melanogaster, oviposition on a substrate containing ethanol enhances fitness; however, little is known about the neural mechanisms mediating this important choice behavior. Here, we characterize the neural modulation of this simple choice and show that distinct subsets of dopaminergic neurons compete to either enhance or inhibit egg-laying preference for ethanol-containing food. Moreover, activity in α'β' neurons of the mushroom body and a subset of ellipsoid body ring neurons (R2) is required for this choice. We propose a model where competing dopaminergic systems modulate oviposition preference to adjust to changes in natural oviposition substrates.
A key step toward understanding a metagenomics data set is the identification of functional sequence elements within it, such as protein coding genes and structural RNAs. Relative to protein coding genes, structural RNAs are more difficult to identify because of their reduced alphabet size, lack of open reading frames, and short length. Infernal is a software package that implements "covariance models" (CMs) for RNA homology search, which harness both sequence and structural conservation when searching for RNA homologs. Thanks to the added statistical signal inherent in the secondary structure conservation of many RNA families, Infernal is more powerful than sequence-only based methods such as BLAST and profile HMMs. Together with the Rfam database of CMs, Infernal is a useful tool for identifying RNAs in metagenomics data sets.
Comprehensive high-resolution structural maps are central to functional exploration and understanding in biology. For the nervous system, in which high resolution and large spatial extent are both needed, such maps are scarce as they challenge data acquisition and analysis capabilities. Here we present for the mouse inner plexiform layer–the main computational neuropil region in the mammalian retina–the dense reconstruction of 950 neurons and their mutual contacts. This was achieved by applying a combination of crowd-sourced manual annotation and machine-learning-based volume segmentation to serial block-face electron microscopy data. We characterize a new type of retinal bipolar interneuron and show that we can subdivide a known type based on connectivity. Circuit motifs that emerge from our data indicate a functional mechanism for a known cellular response in a ganglion cell that detects localized motion, and predict that another ganglion cell is motion sensitive.
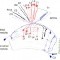
Motion detection is a fundamental neural computation performed by many sensory systems. In the fly, local motion computation is thought to occur within the first two layers of the visual system, the lamina and medulla. We constructed specific genetic driver lines for each of the 12 neuron classes in the lamina. We then depolarized and hyperpolarized each neuron type and quantified fly behavioral responses to a diverse set of motion stimuli. We found that only a small number of lamina output neurons are essential for motion detection, while most neurons serve to sculpt and enhance these feedforward pathways. Two classes of feedback neurons (C2 and C3), and lamina output neurons (L2 and L4), are required for normal detection of directional motion stimuli. Our results reveal a prominent role for feedback and lateral interactions in motion processing and demonstrate that motion-dependent behaviors rely on contributions from nearly all lamina neuron classes.
Cerebellar granule cells constitute the majority of neurons in the brain and are the primary conveyors of sensory and motor-related mossy fiber information to Purkinje cells. The functional capability of the cerebellum hinges on whether individual granule cells receive mossy fiber inputs from multiple precerebellar nuclei or are instead unimodal; this distinction is unresolved. Using cell-type-specific projection mapping with synaptic resolution, we observed the convergence of separate sensory (upper body proprioceptive) and basilar pontine pathways onto individual granule cells and mapped this convergence across cerebellar cortex. These findings inform the long-standing debate about the multimodality of mammalian granule cells and substantiate their associative capacity predicted in the Marr-Albus theory of cerebellar function. We also provide evidence that the convergent basilar pontine pathways carry corollary discharges from upper body motor cortical areas. Such merging of related corollary and sensory streams is a critical component of circuit models of predictive motor control. DOI:http://dx.doi.org/10.7554/eLife.00400.001.
Cell polarization requires increased cellular energy and metabolic output, but how these energetic demands are met by polarizing cells is unclear. To address these issues, we investigated the roles of mitochondrial bioenergetics and autophagy during cell polarization of hepatocytes cultured in a collagen sandwich system. We found that as the hepatocytes begin to polarize, they use oxidative phosphorylation to raise their ATP levels, and this energy production is required for polarization. After the cells are polarized, the hepatocytes shift to become more dependent on glycolysis to produce ATP. Along with this central reliance on oxidative phosphorylation as the main source of ATP production in polarizing cultures, several other metabolic processes are reprogrammed during the time course of polarization. As the cells polarize, mitochondria elongate and mitochondrial membrane potential increases. In addition, lipid droplet abundance decreases over time. These findings suggest that polarizing cells are reliant on fatty acid oxidation, which is supported by pharmacologic inhibition of β-oxidation by etomoxir. Finally, autophagy is up-regulated during cell polarization, with inhibition of autophagy retarding cell polarization. Taken together, our results describe a metabolic shift involving a number of coordinated metabolic pathways that ultimately serve to increase energy production during cell polarization.
The ability to localize proteins precisely within subcellular space is crucial to understanding the functioning of biological systems. Recently, we described a protocol that correlates a precise map of fluorescent fusion proteins localized using three-dimensional super-resolution optical microscopy with the fine ultrastructural context of three-dimensional electron micrographs. While it achieved the difficult simultaneous objectives of high photoactivated fluorophore preservation and ultrastructure preservation, it required a super-resolution optical and specialized electron microscope that is not available to many researchers. We present here a faster and more practical protocol with the advantage of a simpler two-dimensional optical (Photoactivated Localization Microscopy (PALM)) and scanning electron microscope (SEM) system that retains the often mutually exclusive attributes of fluorophore preservation and ultrastructure preservation. As before, cryosections were prepared using the Tokuyasu protocol, but the staining protocol was modified to be amenable for use in a standard SEM without the need for focused ion beam ablation. We show the versatility of this technique by labeling different cellular compartments and structures including mitochondrial nucleoids, peroxisomes, and the nuclear lamina. We also demonstrate simultaneous two-color PALM imaging with correlated electron micrographs. Lastly, this technique can be used with small-molecule dyes as demonstrated with actin labeling using phalloidin conjugated to a caged dye. By retaining the dense protein labeling expected for super-resolution microscopy combined with ultrastructural preservation, simplifying the tools required for correlative microscopy, and expanding the number of useful labels we expect this method to be accessible and valuable to a wide variety of researchers.