Filter
Associated Lab
- Aso Lab (1) Apply Aso Lab filter
- Remove Betzig Lab filter Betzig Lab
- Bock Lab (1) Apply Bock Lab filter
- Clapham Lab (1) Apply Clapham Lab filter
- Fetter Lab (2) Apply Fetter Lab filter
- Harris Lab (2) Apply Harris Lab filter
- Hess Lab (6) Apply Hess Lab filter
- Ji Lab (11) Apply Ji Lab filter
- Lavis Lab (8) Apply Lavis Lab filter
- Lippincott-Schwartz Lab (6) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (6) Apply Liu (Zhe) Lab filter
- Magee Lab (2) Apply Magee Lab filter
- Rubin Lab (1) Apply Rubin Lab filter
- Saalfeld Lab (2) Apply Saalfeld Lab filter
- Schreiter Lab (1) Apply Schreiter Lab filter
- Shroff Lab (9) Apply Shroff Lab filter
- Singer Lab (1) Apply Singer Lab filter
- Svoboda Lab (2) Apply Svoboda Lab filter
- Tjian Lab (4) Apply Tjian Lab filter
- Turner Lab (1) Apply Turner Lab filter
Associated Project Team
Associated Support Team
- Electron Microscopy (2) Apply Electron Microscopy filter
- Integrative Imaging (1) Apply Integrative Imaging filter
- Janelia Experimental Technology (1) Apply Janelia Experimental Technology filter
- Molecular Genomics (1) Apply Molecular Genomics filter
- Primary & iPS Cell Culture (1) Apply Primary & iPS Cell Culture filter
- Project Technical Resources (1) Apply Project Technical Resources filter
- Scientific Computing Software (1) Apply Scientific Computing Software filter
- Viral Tools (1) Apply Viral Tools filter
Publication Date
- 2024 (1) Apply 2024 filter
- 2023 (4) Apply 2023 filter
- 2022 (3) Apply 2022 filter
- 2021 (2) Apply 2021 filter
- 2020 (4) Apply 2020 filter
- 2019 (7) Apply 2019 filter
- 2018 (6) Apply 2018 filter
- 2017 (8) Apply 2017 filter
- 2016 (12) Apply 2016 filter
- 2015 (11) Apply 2015 filter
- 2014 (8) Apply 2014 filter
- 2013 (4) Apply 2013 filter
- 2012 (5) Apply 2012 filter
- 2011 (7) Apply 2011 filter
- 2010 (3) Apply 2010 filter
- 2009 (2) Apply 2009 filter
- 2008 (8) Apply 2008 filter
- 2007 (2) Apply 2007 filter
- 2006 (1) Apply 2006 filter
98 Janelia Publications
Showing 31-40 of 98 resultsCytoskeletal actin dynamics is essential for T cell activation. Here, we show evidence that the binding kinetics of the antigen engaging the T cell receptor influences the nanoscale actin organization and mechanics of the immune synapse. Using an engineered T cell system expressing a specific T cell receptor and stimulated by a range of antigens, we found that the peak force experienced by the T cell receptor during activation was independent of the unbinding kinetics of the stimulating antigen. Conversely, quantification of the actin retrograde flow velocity at the synapse revealed a striking dependence on the antigen unbinding kinetics. These findings suggest that the dynamics of the actin cytoskeleton actively adjusted to normalize the force experienced by the T cell receptor in an antigen-specific manner. Consequently, tuning actin dynamics in response to antigen kinetics may thus be a mechanism that allows T cells to adjust the lengthscale and timescale of T cell receptor signaling.
Sculpting a flat patch of membrane into an endocytic vesicle requires curvature generation on the cell surface, which is the primary function of the endocytosis machinery. Using super-resolved live cell fluorescence imaging, we demonstrate that curvature generation by individual clathrin-coated pits can be detected in real time within cultured cells and tissues of developing organisms. Our analyses demonstrate that the footprint of clathrin coats increases monotonically during the formation of pits at different levels of plasma membrane tension. These findings are only compatible with models that predict curvature generation at the early stages of endocytic clathrin pit formation. We also found that CALM adaptors associated with clathrin plaques form clusters, whereas AP2 distribution is more homogenous. Considering the curvature sensing and driving roles of CALM, we propose that CALM clusters may increase the strain on clathrin lattices locally, eventually giving rise to rupture and subsequent pit completion at the edges of plaques.
In conventional biological imaging, diffraction places a limit on the minimal xy distance at which two marked objects can be discerned. Consequently, resolution of target molecules within cells is typically coarser by two orders of magnitude than the molecular scale at which the proteins are spatially distributed. Photoactivated localization microscopy (PALM) optically resolves selected subsets of protect fluorescent probes within cells at mean separations of <25 nanometers. It involves serial photoactivation and subsequent photobleaching of numerous sparse subsets of photoactivated fluorescent protein molecules. Individual molecules are localized at near molecular resolution by determining their centers of fluorescent emission via a statistical fit of their point-spread-function. The position information from all subsets is then assembled into a super-resolution image, in which individual fluorescent molecules are isolated at high molecular densities. In this paper, some of the limitations for PALM imaging under current experimental conditions are discussed.
Adaptive optics by direct imaging of the wavefront distortions of a laser-induced guide star has long been used in astronomy, and more recently in microscopy to compensate for aberrations in transparent specimens. Here we extend this approach to tissues that strongly scatter visible light by exploiting the reduced scattering of near-infrared guide stars. The method enables in vivo two-photon morphological and functional imaging down to 700 μm inside the mouse brain.
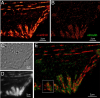
Accurate determination of the relative positions of proteins within localized regions of the cell is essential for understanding their biological function. Although fluorescent fusion proteins are targeted with molecular precision, the position of these genetically expressed reporters is usually known only to the resolution of conventional optics ( approximately 200 nm). Here, we report the use of two-color photoactivated localization microscopy (PALM) to determine the ultrastructural relationship between different proteins fused to spectrally distinct photoactivatable fluorescent proteins (PA-FPs). The nonperturbative incorporation of these endogenous tags facilitates an imaging resolution in whole, fixed cells of approximately 20-30 nm at acquisition times of 5-30 min. We apply the technique to image different pairs of proteins assembled in adhesion complexes, the central attachment points between the cytoskeleton and the substrate in migrating cells. For several pairs, we find that proteins that seem colocalized when viewed by conventional optics are resolved as distinct interlocking nano-aggregates when imaged via PALM. The simplicity, minimal invasiveness, resolution, and speed of the technique all suggest its potential to directly visualize molecular interactions within cellular structures at the nanometer scale.
Commentary: Identifies the photoactivatable fluorescent proteins (PA-FPs) Dronpa and PS-CFP2 as green partners to orange-red PA-FPs such as Kaede and Eos for dual color PALM imaging. Very low crosstalk is demonstrated between the two color channels. Furthermore, since the probes are genetically expressed, they are closely bound to their target proteins and exhibit zero non-specific background. All these properties are essential to unambiguously identify regions of co-localization or separate compartmentalization at the nanoscale, as demonstrated in the examples here.
Cells in the brain act as components of extended networks. Therefore, to understand neurobiological processes in a physiological context, it is essential to study them in vivo. Super-resolution microscopy has spatial resolution beyond the diffraction limit, thus promising to provide structural and functional insights that are not accessible with conventional microscopy. However, to apply it to in vivo brain imaging, we must address the challenges of 3D imaging in an optically heterogeneous tissue that is constantly in motion. We optimized image acquisition and reconstruction to combat sample motion and applied adaptive optics to correcting sample-induced optical aberrations in super-resolution structured illumination microscopy (SIM) in vivo. We imaged the brains of live zebrafish larvae and mice and observed the dynamics of dendrites and dendritic spines at nanoscale resolution.
The development and maintenance of tissues requires collective cell movement, during which neighbouring cells coordinate the polarity of their migration machineries. Here, we ask how polarity signals are transmitted from one cell to another across symmetrical cadherin junctions, during collective migration. We demonstrate that collectively migrating endothelial cells have polarized VE-cadherin-rich membrane protrusions, ‘cadherin fingers’, which leading cells extend from their rear and follower cells engulf at their front, thereby generating opposite membrane curvatures and asymmetric recruitment of curvature-sensing proteins. In follower cells, engulfment of cadherin fingers occurs along with the formation of a lamellipodia-like zone with low actomyosin contractility, and requires VE-cadherin/catenin complexes and Arp2/3-driven actin polymerization. Lateral accumulation of cadherin fingers in follower cells precedes turning, and increased actomyosin contractility can initiate cadherin finger extension as well as engulfment by a neighbouring cell, to promote follower behaviour. We propose that cadherin fingers serve as guidance cues that direct collective cell migration.
Super-resolution fluorescence microscopy is distinct among nanoscale imaging tools in its ability to image protein dynamics in living cells. Structured illumination microscopy (SIM) stands out in this regard because of its high speed and low illumination intensities, but typically offers only a twofold resolution gain. We extended the resolution of live-cell SIM through two approaches: ultrahigh numerical aperture SIM at 84-nanometer lateral resolution for more than 100 multicolor frames, and nonlinear SIM with patterned activation at 45- to 62-nanometer resolution for approximately 20 to 40 frames. We applied these approaches to image dynamics near the plasma membrane of spatially resolved assemblies of clathrin and caveolin, Rab5a in early endosomes, and α-actinin, often in relationship to cortical actin. In addition, we examined mitochondria, actin, and the Golgi apparatus dynamics in three dimensions.
Despite the apparent simplicity of the xanthene fluorophores, the preparation of caged derivatives with free carboxy groups remains a synthetic challenge. A straightforward and flexible strategy for preparing rhodamine and fluorescein derivatives was developed using reduced, “leuco” intermediates.
Gap junctions (GJs) represent connexin-rich membrane domains that connect interiors of adjoining cells in mammalian tissues. How fast GJs can respond to bacterial pathogens has not been known previously. Using Bessel beam plane illumination and confocal spinning disk microscopy, we found fast ( 500 ms) formation of connexin-depleted regions (CDRs) inside GJ plaques between cells exposed to AB5 toxins. CDR formation appears as a fast redistribution of connexin channels within GJ plaques with minor changes in outline or geometry. CDR formation does not depend on membrane trafficking or submembrane cytoskeleton and has no effect on GJ conductance. However, CDR responses depend on membrane lipids, can be modified by cholesterol-clustering agents and extracellular K(+) ion concentration, and influence cAMP signaling. The CDR response of GJ plaques to bacterial toxins is a phenomenon observed for all tested connexin isoforms. Through signaling, the CDR response may enable cells to sense exposure to AB5 toxins. CDR formation may reflect lipid-phase separation events in the biological membrane of the GJ plaque, leading to increased connexin packing and lipid reorganization. Our data demonstrate very fast dynamics (in the millisecond-to-second range) within GJ plaques, which previously were considered to be relatively stable, long-lived structures.