Filter
Associated Lab
- Aso Lab (2) Apply Aso Lab filter
- Betzig Lab (6) Apply Betzig Lab filter
- Card Lab (1) Apply Card Lab filter
- Clapham Lab (2) Apply Clapham Lab filter
- Fetter Lab (5) Apply Fetter Lab filter
- Funke Lab (5) Apply Funke Lab filter
- Harris Lab (2) Apply Harris Lab filter
- Remove Hess Lab filter Hess Lab
- Jayaraman Lab (1) Apply Jayaraman Lab filter
- Lavis Lab (2) Apply Lavis Lab filter
- Lee (Albert) Lab (1) Apply Lee (Albert) Lab filter
- Lippincott-Schwartz Lab (12) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (2) Apply Liu (Zhe) Lab filter
- Looger Lab (2) Apply Looger Lab filter
- Rubin Lab (5) Apply Rubin Lab filter
- Saalfeld Lab (8) Apply Saalfeld Lab filter
- Scheffer Lab (10) Apply Scheffer Lab filter
- Shroff Lab (1) Apply Shroff Lab filter
- Turner Lab (1) Apply Turner Lab filter
Associated Project Team
Associated Support Team
- Anatomy and Histology (1) Apply Anatomy and Histology filter
- Cryo-Electron Microscopy (1) Apply Cryo-Electron Microscopy filter
- Electron Microscopy (4) Apply Electron Microscopy filter
- Janelia Experimental Technology (3) Apply Janelia Experimental Technology filter
- Molecular Genomics (1) Apply Molecular Genomics filter
- Primary & iPS Cell Culture (2) Apply Primary & iPS Cell Culture filter
- Project Technical Resources (2) Apply Project Technical Resources filter
- Scientific Computing Software (5) Apply Scientific Computing Software filter
- Scientific Computing Systems (1) Apply Scientific Computing Systems filter
- Viral Tools (1) Apply Viral Tools filter
Publication Date
- 2024 (1) Apply 2024 filter
- 2023 (6) Apply 2023 filter
- 2022 (7) Apply 2022 filter
- 2021 (5) Apply 2021 filter
- 2020 (5) Apply 2020 filter
- 2019 (5) Apply 2019 filter
- 2018 (3) Apply 2018 filter
- 2017 (4) Apply 2017 filter
- 2016 (2) Apply 2016 filter
- 2015 (7) Apply 2015 filter
- 2014 (5) Apply 2014 filter
- 2013 (2) Apply 2013 filter
- 2012 (3) Apply 2012 filter
- 2011 (2) Apply 2011 filter
- 2010 (2) Apply 2010 filter
- 2009 (2) Apply 2009 filter
- 2008 (2) Apply 2008 filter
- 2007 (1) Apply 2007 filter
- 2006 (1) Apply 2006 filter
65 Janelia Publications
Showing 31-40 of 65 resultsWe demonstrate gas cluster ion beam scanning electron microscopy (SEM), in which wide-area ion milling is performed on a series of thick tissue sections. This three-dimensional electron microscopy technique acquires datasets with <10 nm isotropic resolution of each section, and these can then be stitched together to span the sectioned volume. Incorporating gas cluster ion beam SEM into existing single-beam and multibeam SEM workflows should be straightforward, increasing reliability while improving z resolution by a factor of three or more.
Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) generates 3D datasets optimally suited for segmentation of cell ultrastructure and automated connectome tracing but is limited to small fields of view and is therefore incompatible with the new generation of ultrafast multibeam SEMs. In contrast, section-based techniques are multibeam-compatible but are limited in z-resolution making automatic segmentation of cellular ultrastructure difficult. Here we demonstrate a novel 3D electron microscopy technique, Gas Cluster Ion Beam SEM (GCIB-SEM), in which top-down, wide-area ion milling is performed on a series of thick sections, acquiring < 10 nm isotropic datasets of each which are then stitched together to span the full sectioned volume. Based on our results, incorporating GCIB-SEM into existing single beam and multibeam SEM workflows should be straightforward and should dramatically increase reliability while simultaneously improving z-resolution by a factor of 3 or more.
View Publication PageThe challenge of recovering the topology of massive neuronal circuits can potentially be met by high throughput Electron Microscopy (EM) imagery. Segmenting a 3-dimensional stack of EM images into the individual neurons is difficult, due to the low depth-resolution in existing high-throughput EM technology, such as serial section Transmission EM (ssTEM). In this paper we propose methods for detecting the high resolution locations of membranes from low depth-resolution images. We approach this problem using both a method that learns a discriminative, over-complete dictionary and a kernel SVM. We test this approach on tomographic sections produced in simulations from high resolution Focused Ion Beam (FIB) images and on low depth-resolution images acquired with ssTEM and evaluate our results by comparing it to manual labeling of this data.
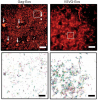
We combined photoactivated localization microscopy (PALM) with live-cell single-particle tracking to create a new method termed sptPALM. We created spatially resolved maps of single-molecule motions by imaging the membrane proteins Gag and VSVG, and obtained several orders of magnitude more trajectories per cell than traditional single-particle tracking enables. By probing distinct subsets of molecules, sptPALM can provide insight into the origins of spatial and temporal heterogeneities in membranes.
Commentary: As a stepping stone to true live cell PALM (see above), our collaborator Jennifer Lippincott-Schwartz suggested using the sparse photoactivation principle of PALM to track the nanoscale motion of thousands of individual molecules within a single living cell. Termed single particle tracking PALM (sptPALM), Jennifer’s postdocs Suliana Manley and Jen Gillette used the method in our PALM rig to create spatially resolved maps of diffusion rates in the plasma membrane of live cells. sptPALM is a powerful tool to study the active cytoskeletal or passive diffusional transport of individual molecules with far more measurements per cell than is possible without sparse photoactivation.
The automated tape-collecting ultramicrotome (ATUM) makes it possible to collect large numbers of ultrathin sections quickly-the equivalent of a petabyte of high resolution images each day. However, even high throughput image acquisition strategies generate images far more slowly (at present ~1 terabyte per day). We therefore developed WaferMapper, a software package that takes a multi-resolution approach to mapping and imaging select regions within a library of ultrathin sections. This automated method selects and directs imaging of corresponding regions within each section of an ultrathin section library (UTSL) that may contain many thousands of sections. Using WaferMapper, it is possible to map thousands of tissue sections at low resolution and target multiple points of interest for high resolution imaging based on anatomical landmarks. The program can also be used to expand previously imaged regions, acquire data under different imaging conditions, or re-image after additional tissue treatments.
Many biomolecules in cells can be visualized with high sensitivity and specificity by fluorescence microscopy. However, the resolution of conventional light microscopy is limited by diffraction to ~200-250nm laterally and >500nm axially. Here, we describe superresolution methods based on single-molecule localization analysis of photoswitchable fluorophores (PALM: photoactivated localization microscopy) as well as our recent three-dimensional (3D) method (iPALM: interferometric PALM) that allows imaging with a resolution better than 20nm in all three dimensions. Considerations for their implementations, applications to multicolor imaging, and a recent development that extend the imaging depth of iPALM to ~750nm are discussed. As the spatial resolution of superresolution fluorescence microscopy converges with that of electron microscopy (EM), direct imaging of the same specimen using both approaches becomes feasible. This could be particularly useful for cross validation of experiments, and thus, we also describe recent methods that were developed for correlative superresolution fluorescence and EM.

We introduce a method for optically imaging intracellular proteins at nanometer spatial resolution. Numerous sparse subsets of photoactivatable fluorescent protein molecules were activated, localized (to approximately 2 to 25 nanometers), and then bleached. The aggregate position information from all subsets was then assembled into a superresolution image. We used this method–termed photoactivated localization microscopy–to image specific target proteins in thin sections of lysosomes and mitochondria; in fixed whole cells, we imaged vinculin at focal adhesions, actin within a lamellipodium, and the distribution of the retroviral protein Gag at the plasma membrane.
Commentary: The original PALM paper by myself and my friend and co-inventor Harald Hess, spanning the before- and after-HHMI eras. Submitted and publicly presented months before other publications in the same year, the lessons of the paper remain widely misunderstood: 1) localization precision is not resolution; 2) the ability to resolve a few molecules by the Rayleigh criterion in a diffraction limited region (DLR) does not imply the ability to resolve structures of arbitrary complexity at the same scale; 3) true resolution well beyond the Abbe limit requires the ability to isolate and localize hundreds or thousands of molecules in one DLR; and 4) certain photoactivatable fluorescent proteins (PA-FPs) and caged dyes can be isolated and precisely localized at such densities; yielding true resolution down to 20 nm. The molecular densities we demonstrate (105 molecules/m2) are more than two orders of magnitude greater than in later papers that year (implying ten-fold better true resolution) – indeed, these papers demonstrate densities only comparable to earlier spectral or photobleaching based isolation methods. We validate our claims by correlative electron microscopy, and demonstrate the outstanding advantages of PA-FPs for superresolution microscopy: minimally perturbative sample preparation; high labeling densities; close binding to molecular targets; and zero non-specific background.
The molecular mechanism responsible for capturing, sorting and retrieving vesicle membrane proteins following triggered exocytosis is not understood. Here we image the post-fusion release and then capture of a vesicle membrane protein, the vesicular acetylcholine transporter, from single vesicles in living neuroendocrine cells. We combine these measurements with super-resolution interferometric photo-activation localization microscopy and electron microscopy, and modelling to map the nanometer-scale topography and architecture of the structures responsible for the transporter’s capture following exocytosis. We show that after exocytosis, the transporter rapidly diffuses into the plasma membrane, but most travels only a short distance before it is locally captured over a dense network of membrane-resident clathrin-coated structures. We propose that the extreme density of these structures acts as a short-range diffusion trap. They quickly sequester diffusing vesicle material and limit its spread across the membrane. This system could provide a means for clathrin-mediated endocytosis to quickly recycle vesicle proteins in highly excitable cells.
The endoplasmic reticulum (ER) is an expansive, membrane-enclosed organelle that plays crucial roles in numerous cellular functions. We used emerging superresolution imaging technologies to clarify the morphology and dynamics of the peripheral ER, which contacts and modulates most other intracellular organelles. Peripheral components of the ER have classically been described as comprising both tubules and flat sheets. We show that this system consists almost exclusively of tubules at varying densities, including structures that we term ER matrices. Conventional optical imaging technologies had led to misidentification of these structures as sheets because of the dense clustering of tubular junctions and a previously uncharacterized rapid form of ER motion. The existence of ER matrices explains previous confounding evidence that had indicated the occurrence of ER “sheet” proliferation after overexpression of tubular junction–forming proteins.